Reports: ND152737-ND1: Catalytic Asymmetric Carbon-Carbon Bond Formation with Fluoroenolates Generated Detylated Prenucleophiles
Christian Wolf, Georgetown University
Chiral
organofluorines have become increasingly attractive synthetic and medicinal targets
because they often afford superior lipophilicity, bioavailability and
resistance to metabolic degradation compared to the nonfluorinated parent
compounds. The incorporation of a difluoromethylene group is particularly
interesting because the high electronegativity of fluorine leads to increased
dipole moments, conformational changes, carbonyl hydration and reduced pKa
values of adjacent functionalities. The catalytic asymmetric formation of
chiral compounds with a neighboring difluoromethylene group has remained a
major problem, mostly because of the difficulty with forming difluoroenolates under
conditions suitable for subsequent stereocontrolled C-C bond formation.
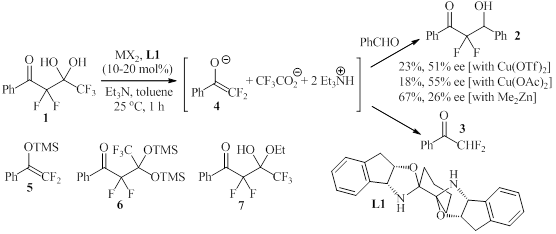
Scheme 1. Initial analysis of the catalytic
difluoromethylation of aldehydes.To address the challenges outlined
above, we investigated the possibility of catalytic enantioselective aldol
reactions using aldehydes and difluoroenolates generated in situ from gem-diol
1 and its derivatives (Scheme 1). During initial NMR and ReactIR
analysis of the effect of additives, base, solvent and other parameters on this
reaction we found that catalytic bond cleavage of 1 and subsequent C-C
bond formation toward 2 is possible in the presence of 10-20 mol% of
magnesium, copper and zinc salts. However, formation of substantial amounts of
by-product 3 was observed. The testing of alternative starting materials
5-7 did not improve results. After screening of several reaction
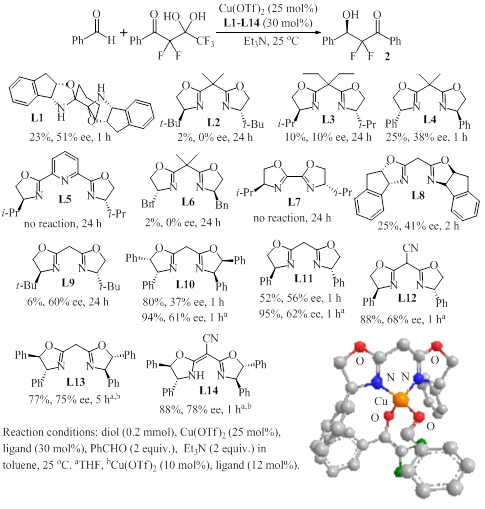
conditions, we decided to apply several bisoxazoline ligands to our general
reaction protocol. While the results obtained in the presence of catalytic
amounts of copper(II) triflate and L2-L9 varied considerably, L10-L12
showed remarkable promise (Figure 1). We were very pleased to find that 2
is formed in almost quantitative yields and 61-68% ee within 1 h when these
ligands are employed in THF. Our screening results revealed that anionic
ligands exhibiting a delocalized negative charge in the backbone, in particular
semicorrin L12, allow completion of the reaction within 1 h. We
rationalized that such a catalytically more active catalyst would enable us to
perform the reaction at lower temperature to improve the enantioselectivity
while the competing protonation of the enolate to by-product 3 would
remain relatively slow and thus not affect yields. We therefore further
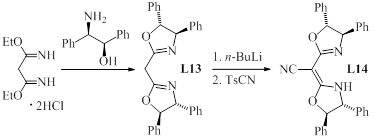
modified the ligand structure and prepared L13 and L14 (Scheme
2). A change of the relative configuration of the adjacent phenyl rings in L10
to anti-derivative L13 improved the enantioselectivity and
introduction of a cyano group to the methylene bridge further increased
catalytic activity and yield. With L14 in hand, we were able to decrease
the catalyst loading to 5 mol% and obtained 2 as well as analogues 8-10
in up to 98% yield and 92% ee using 1.2 equivalents of benzaldehyde at 10 oC
(Scheme 3).
Figure 1. Chiral ligand screening results and
MOPAC computation of the loaded catalyst (hydrogens are omitted for clarity).
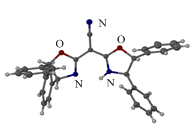
Scheme 2. Synthesis and single crystal
structure of L14.
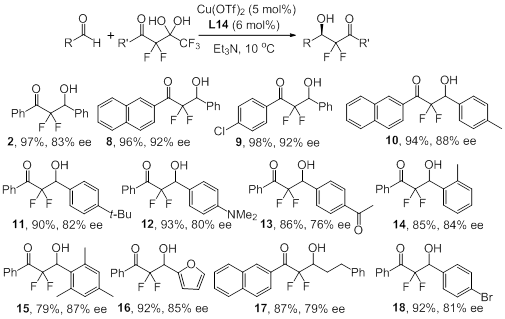
Scheme 3. Scope of the enantioselective aldol
reaction (selected ecxample).
We then applied a wide range of
aldehydes to evaluate the reaction scope. In general, high yields and enantiomeric
excess were obtained with electron-rich and electron-deficient substrates.
Importantly, this method tolerates the presence of amine, ketone, ester and
other functional groups, and the reaction with sterically hindered aldehydes
gave 14 and 15 with 79-85% yield and 84-87% ee. Our procedure
provides unprecedented asymmetric access to important chiral building blocks,
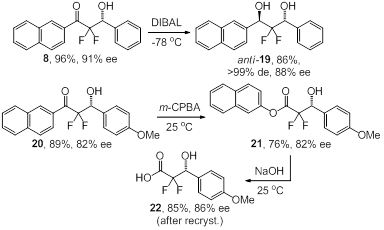
including 2,2-difluoro-1,3-propanols. For example, the reduction of 8
with DIBAL produces anti-19 in high yields and with excellent
diastereoselectivity (Scheme 4). This establishes a convenient entry to C1-
and C2-symmetric anti-1,3-diols motifs which appear in many
natural products and are of great pharmaceutical interest. Alternatively, we
realized that the Baeyer-Villiger oxidation followed by mild hydrolysis affords
the first asymmetric route to 2,2-difluoro-3-hydroxy carboxylic acids which are
crucial precursors of several enzyme inhibitors and other biologically active
compounds. Oxidation of 20 with m-CPBA to the corresponding
2-naphthoate 21 and basic hydrolysis gave 2,2-difluoro-3-hydroxy acid 22
in 85% yield and 86% ee.
Scheme 4. Formation of a representative anti-2,2-difluoro-1,3-diol
and a 2,2-difluoro-3-hydroxy acid.
In conclusion, we have introduced a
practical method for the catalytic enantioselective addition of
α,α-difluoroenolates to aldehydes using 5 mol% of copper(II) triflate
and a new bisoxazoline ligand which can be easily prepared in two steps. High
yields and enantiomeric excess were obtained with a wide range of
prenucleophiles and aldehydes, including aliphatic substrates, under mild conditions
and in short reaction times. The value of this reaction is highlighted by the
stereoselective reduction and Baeyer-Villiger oxidation of alpha,alpha-difluoro-beta-hydroxy
ketones to an anti-2,2-difluoro-1,3-propanol and a
2,2-difluoro-3-hydroxy carboxylic acid, respectively.
 printer friendly
printer friendly