Reports: ND653161-ND6: A Comprehensive Molecular-Based Study of the Stability of Clathrate Hydrates
 printer friendly
printer friendlyThe aim of this work is the formulation of methods for calculation of free energies of crystalline solids, and their application to calculation of the phase diagram of model clathrate hydrates, considering a variety of guest molecules. Progress on the project since the initial award has proceeded in several ways.
First, we have established the modeling framework with details appropriate to the hydrate clathrate systems. This includes: setting up the molecular force fields; coding the nominal crystal structures; coding algorithms for implementing the proton-disordered structures satisfying the Bernal-Fowler rules with near-zero dipole moment; and implementing our lattice-dynamics and molecular dynamics codes for the system.
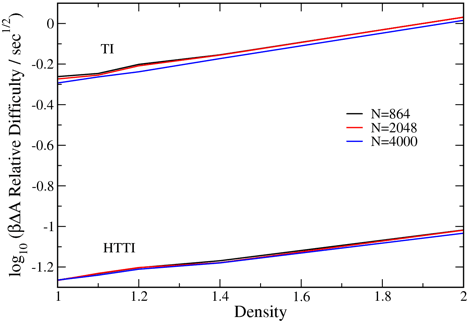
Second, we have made good progress in developing and testing new free-energy methods that will be applied to the models in the next grant period. We have in fact invented another method that complements the harmonically targeted temperature perturbation (HTTP) technique described in our proposal and published previously. The new method is called harmonically targeted thermodynamic integration (HTTI). This method is a thermodynamic integration (TI) counterpart to the HTTP method discussed. TI in general provides the free energy in the form of an integral, wherein the integrand is the derivative of the free energy with respect to the integration variable (in our case the temperature), and is given via an ensemble average evaluated by molecular simulation. We enhance TI for solid-state applications by again invoking the harmonic character of the crystal (as we did with HTTP), such that the quantity computed by simulation expresses purely the anharmonic contribution to the TI integrand; the harmonic contribution again is easily evaluated analytically and added in afterward. We have tested this method first in application to a spherical molecular model, so that we can avoid dealing with rotational degrees of freedom. Specifically, we performed calculations for an extended Finnis-Sinclair (EFS) potential for tungsten (W). This model is given as a conventional central pair potential plus a multibody embedding function expressed in terms of electronic density obtained by summing over the individual atoms, and as such, it presents a slightly more complicated and challenging model than provided by the usual Lennard-Jones system (which we have also examined). The advantage gained by this harmonically-targeted thermodynamic integration (HTTI) approach in application to the EFS model for W is shown here:
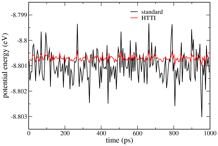
The figure on the left (which is for 300K, 128 atoms)
demonstrates the enhanced precision of HTTI relative to standard TI. These are
traces of short block averages the TI integrand from HTTI (red) and the
standard integrand (black), through the course of a molecular dynamics (MD)
simulation. The standard method clearly suffers from fluctuations in the
harmonic contributions to the integrand. The net outcome is that the HTTI
method provides results to the same precision as standard TI but with computational
effort that is 5 to 50 times less (depending on temperature – greater
advantage for HTTI is found at low temperature, where the harmonic
contributions dominate the integrand). Also notable in the figure is a slight vertical
shift in the traces relative to each other, which is related to the better
handling of finite-size effects in the HTTI calculation. This feature is better
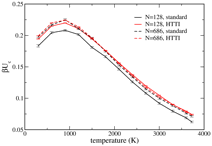
seen in the figure on the right, which shows averages of the integrand for TI
and for HTTI, each evaluated for two system sizes. Again it is clear that the
HTTI integrand (red), which includes only anharmonic contributions, is
practically independent of system size, whereas the TI integrand (black) has a
noticeable dependence.
Third, we have developed a framework for implementation of
HTTP and HTTI to non-spherical molecules, which of course is important to the present
project and its application to water molecules. In the process we have developed
a novel way to represent orientations, which appears to show some advantages
when applied in connection with the HTTP/HTTI methods being developed here;
they are also convenient for the lattice dynamics calculations that are the
starting point for the overall approach. In this treatment, working with the
well-known inertia-weighted dynamical matrices is avoided if the sole purpose
is to compute the free energy. Lattice dynamics calculations using this framework
have been applied to different proton-disordered clathrate hydrate crystal structures
(sI, sII and sH). The TIP4P potential was used, where the long-range Coulomb
interactions are treated using the Ewald sum technique while a lattice sum is
used for van der Waals 12-6 Lennard-Jones potential. The free-energy results
agree well with previous work.
In summary, our progress over the grant period so far has
put us in a good position to move ahead now with at least some of the specific
calculations described in our proposal. We expect that additional development
will still be needed for the proposed consideration of nuclear quantum effects
as well.