Reports: DNI653049-DNI6: Characterization of Micelle Formation and Stability Under Extreme Conditions
 printer friendly
printer friendlyTo obtain the correct balance of hydrophobic/hydrophilic driving forces, we have initially parameterized a heteropolymer model with only two different coarse-grained bead types, polar (P) or hydrophobic (H), to exhibit cold and pressure denaturation. Polar interactions (for both polymer and water) are modeled using a three-body anisotropic potential that effectively models hydrogen bonding by imposing a penalty on nontetrahedral configurations. Interactions involving hydrophobic beads are modeled using a 9-6 Lennard-Jones potential. The results of this work have been published in Physical Review Letters1. We designed foldable primarily hydrophobic sequences with fixed polar content in water at physiological conditions, which has demonstrated a temperature-pressure dependence of conformational stability similar to biological proteins. Inherent helicity emerges as a result of local polar-polar interaction in the sequences that mimic a-helices. The enhancement of tetrahedrality of the hydrophilic hydration shell at cold temperature and high pressure reduces the interactions between water and polar residues. Such reduction promotes polar-polar interactions at high pressure and cold temperature, which is similar to results obtained in vacuum simulations. Consequently at these conditions a polar core is observed. While changes in hydrophobic solvation are the driving forces underlying cold- and pressure-induced unfolding, we observed that changes in hydrophilic hydration have a crucial contribution toward shaping the conformational landscape at those nonphysiological conditions. These observations are in agreement to the experimental observation that for nonionic surfactants there is a collapse of the hydrophilic part of the surfactant at high pressures2. Also an increase in helicity has been observed at low temperatures and high pressure due to an enhancement of polar-polar interactions where hydrophobic interactions are weak at those conditions. This is in agreement with the experimental observation that an emergence of helical structure is observed in ubiquitin and polyalanine at low temperature and high pressure3-5.
Once we observed the right hydrophobic/hydrophilic trends with changes in temperature and pressure we have moved towards the modeling of -alkyl polyoxyethelyne ether (CnEm). Experimentally it has been shown that this type of surfactant can form a variety of microstructures, which depends on the copolymer length, in water ranging from simple micelles to complex mesophases. Figure 1 shows a cartoon of how the coarse-grained is done in C12E6. Each heavy atom is coarse-grained with a single bead.
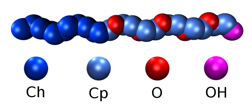
Figure 1: C12E5represented by 4 CG beads.
The carbons that belong to the alkyl tail (Ch) have different properties than the ones in the polar regions (Cp), therefore we have used different bead types that has a different LJ interaction with the solvent. Also the beads that model the ester oxygens (O) do not exhibit hydrogen-bonding capabilities between themselves, whereas the bead that encompasses the hydroxy group (OH) does. As we did for the heteropolymer model, by using a three body anisotropic potential we have mimicked in an effective way the hydrogen bonding between the O and OH and the water solvent, and between the OH groups. Each water molecule is modeled as a single bead that interacts between themselves with a three body potential. We observe a speedup of 100 with our coarse-grained model when compared to atomistic simulations.
We did a further parametrization from the heteropolymer model by perfoming simulations with two dimers. As shown in Figure 2, the agreement between our CG model and all-atom simulations for the radial distribution function between the surfactant groups and the water molecules is excellent. Thus, capturing the right solvation properties. Self-assembly simulations of 160 surfactants with a concentration above CMC yield the formation of two micelles in equilibrium at 350K and 1atm. Figure 3b depicts the time evolution of the fraction of free monomers, showing that the system reaches equilibrium at 180ns. Figure 3a shows the time evolution of the size of the largest 2 clusters with a size of 60 and 52 surfactants. A cartoon of a typical micelle at 350K and 1atm is shown in Figure 4. Blue correspond to the alkyl tail and red to the ether domain. Clearly the micelle has a hydrophobic core with the ether domain facing the solvent. At a temperature of 600K, there is no formation of micelles (results not shown). We are currently performing simulations at various temperatures and different surfactant lengths to explore morphological changes and how solvation effects drive those changes.
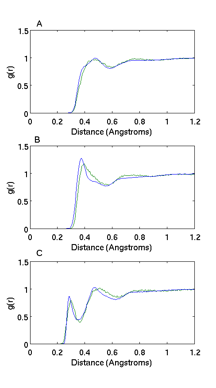
Figure 2: Radial distribution function of water around A) carbon in alkyl domain (Ch), B) carbon in the ether domain (Cp) and C) oxygen in the ether domain. Data from all-atom simulation is in blue, while data from the CG model is in green.
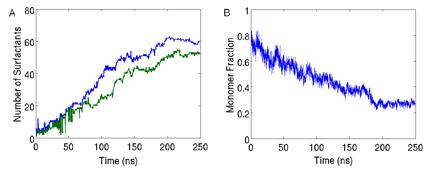
Figure 3: A) Time evolution of the number of surfactants in the two largest clusters. B) Time evolution of the fraction of the total number of surfactants that are not in a cluster.
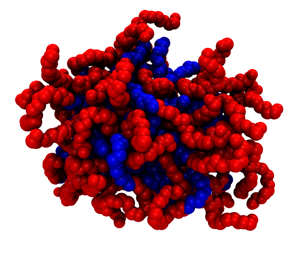
Figure 4: Micelle containing 60 C12E5molecules. The alkyl domain is in blue and the ether is in red.
References:
1. Matysiak S., Das P., Phys. Rev. Lett., 111, 058103 (2013).
2. Lesemann M., Nathan H., DiNoia T. P., Kirby C.F., McHugh M. A., van Zanten J. H., Paulaitis M.E., Ind. Eng. Chem. Res., 42, 6465 (2003).
3 Herberhold H, Winter R., , Biochemistry, 41, 2396 (2002)
4. Vajpai N., Nisius L.,Wiktor M., Grzesiek S., Proc. Natl. Acad. Sci., 110, E368 (2013)
5 Yoshidome T., Harano Y., Kinoshita M., Phys. Rev. E, 79, 011912 (2009)










