Reports: DNI353298-DNI3: Harnessing Zwitterions for Late-Transition-Metal Catalysis
 printer friendly
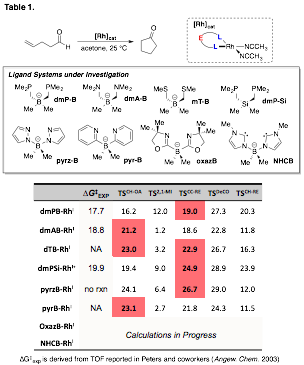
printer friendlyA long-standing problem in catalytic hydroacylation is competitive decarbonylation. A DFT reaction coordinate analysis of intramolecular hydroacylation using a promising catalyst, zwitterionic rhodium(I)-bisphosphinoborate, 1, is nearing completion. The complete reaction coordinate was found to be in good agreement with experiment (Figure 1). The important kinetic events (ie, hydroacylation and decarbonylation transition states) were further analyzed for a variety of L2-borate ligands (Table 1). While electron-rich dmP-B ligand has an expected turnover-limiting step (TLS) of C-C reductive elimination (TSCC-RE), less electron rich ligands invert to a predicted TLS of C-H oxidative addition (TSCH-OA) but also have more kinetically competitive decarbonylation pathways. These calculations will serve as the foundation for ongoing experimental catalyst discovery for hydroacylation.

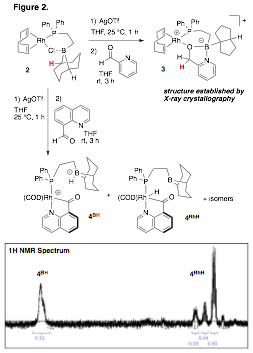
An alternative proposed strategy to disfavor decarbonylation uses a secondary sphere borane from a phosphino-borane ligand (R2P-CH2-CH2-BR2) to stabilize the acyl ligand resulting from C-H oxidative addition at rhodium(I). While preliminary catalytic studies have not born fruit, we are characterizing the reactivity of directed substrates (2-formylpyridine and 9-formylquinolyn) to establish the viability of the proposed strategy (Figure 2). Interestingly, 2-formylpyridine had expected reactivity with complex 3 being isolated and characterized. Initial evidence suggests that it arises from a transfer hydrogenation of aldehyde from an initial C–H oxidative addition from a methine on the 9-BBN ring. 9-formylquinolyn reacts to yield a mixture of rhodium-hydride (4RhH) and zwitterionic hydrido-borate (4BH) products (Figure 2). 11B NMR spectroscopy has been inconclusive as to the borane interactions in the 4RhH species. Future work will compare the mixture of 4 with a non-boron containing analog to establish decarbonylation kinetics.
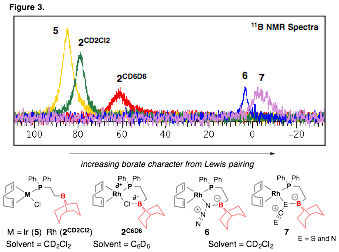
It became apparent in our previous studies of aldehyde oxidative addition that characterization of boron-ligand interactions by 11B NMR spectroscopy will be crucial to understand boron's stabilizing role. We have begun to identify solvent and counter-ion ligand interactions with boron (Figure 3). While these interactions have been described in a general manner in the literature, we are currently carrying out studies on series of compounds (eg., F, Cl, Br, I) and solvents (eg., % fraction CD2Cl2 in C6D6) in order to establish chemical shift and/or line width correlations with other system parameters (eg., ligand ionicity and solvent polarity).
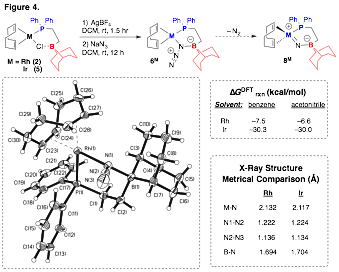
We have also begun to explore whether small molecules can be activated using a second-sphere borane moiety. For example, we prepared and characterized azide complexes of RhI and IrI (6M) in which the azide moiety features short N1 to Rh and B interactions, respectively (Figure 4). Importantly, the N3 bonding is polarized by almost 1, suggesting that loss of N2 should be facile. Indeed, upon heating 6Ir in toluene under a slightly reduced pressure, a new complex is formed cleanly but has yet eluded isolation and characterization. DFT calculations support the viability of a formal zwitterionic metal-nitride intermediate 8M upon octahedral-to-tetrahedral ligand sphere rearrangement, yielding a species that will be expected to be highly reactive toward ligand functionality in the complex. Ancillary ligands other than COD (eg., bipyridine) are currently being explored as an oxidatively-stable alternative to give us a realistic opportunity at isolating a potential formal Group IX nitride species.
Phosphino-borane ligands have not been explored widely for catalytic reactions. Therefore, we sought to establish their viability for a benchmark reaction–hydroboration. Crudden and coworkers (J. Am. Chem. Soc. 2009) recently demonstrated the significant beneficial influence of Lewis acidic boron, providing further motive for our study. While studies of styrene derivatives showed little significance from a methodology perspective, we did identify interesting features when exploring the stoichiometric reactivity of complex 2Rh with pinacolborane. Specifically, we have evidence for the formation of pinacolborenium ion and a hydride bridged between Rh and ligand 9BBN. This intermediate is a critical catalytic intermediate proposed in Crudden's studies and therefore further characterization is likely to yield useful insights into her system's catalytic mechanism.
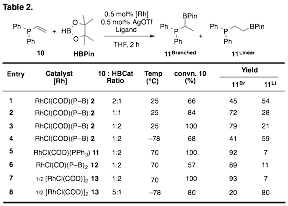
In screening different substrates with phophino-borane-ligated rhodium catalysts, we found that mixtures of branched and linear hydroboration products of diphenylvinylphosphine, 10, were obtained. Further screening revealed that simple pre-catalyst [Rh(COD)Cl]2, 13, can provide up to 13:1 branched product and 1:4 linear product when the reaction is run at 70°C and –78°C, respectively. To our knowledge this is the first example of a single catalyst that provides each regioisomeric product with high selectivity. The temperature dependent selectivity hints at the possibility that Lewis pair interactions may be important thus warranting further investigations. Given the versatility of alkyl-BPin in a variety of transmetalative cross coupling processes, we believe that this procedure will find significant utility in the catalysis and synthetic communities.
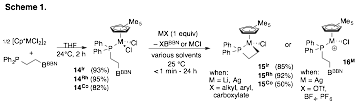
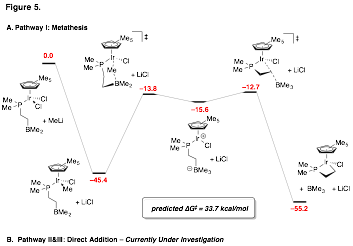
We have also begun studying trivalent Group IX metal complexes featuring the phosphino-borane ligand. In particular we were interested in exploring the dynamical behavior of strong anionic donor ligands (eg., hydride, alkyl, aryl) in the presence of outer-sphere borane. Surprisingly when metathesis conditions were employed with anionic, coordinating metal salts, we observed near selective formation of a novel metallacyclic product, 15M, for all Group IX complexes. In contrast anionic, non-coordinating metal salts give rise to coordinately unsaturated metal complexes, 16M (Scheme 1). The presumed metathesis/zwitterion mechanism for the formation of a model metallacycle of 15Ir was explored using DFT methods (Figure 5). We found that the barrier for zwitterion formation via methyl transfer was much too high to be consistent with the near instantaneous formation of metallacycle observed by experiment. A DFT study is now underway to establish the viability a different varients of a mechanism in which direct addition of nucleophilic salt to the borane center induces metallacycle formation. We expect that this work will be important when considering the reactivity of this ambiphilic ligand class in the presence of electrophilic late transition metals.