Reports: DNI351754-DNI3: Prodigiosin-Based Platforms in Metalloradical Complexes for Dioxygen Activation and Oxidative Catalysis
 printer friendly
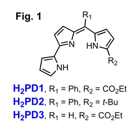
printer friendlyWe developed a modular synthesis of meso-aryl pyrrolyldipyrrin scaffolds H2PD1 and H2PD2 (Fig. 1), which were designed to present enhanced coordinating ability. The alpha-ester substitution in H2PD1 was expected to provide not only an additional electron-withdrawing group but also a neutral ligand for metal coordination (see below). The H2PD2 scaffold carrying the bulky, non-coordinating, electron-donating tert-butyl group was selected for an informative comparison on electronic and coordinating effects on the meso-aryl pyrrolyldipyrrin platform. In addition, the alpha-ester analog H2PD3 (Fig. 1), lacking a meso-aryl substituent, was prepared by adaptation of previously reported syntheses of prodigiosin analogs.2,3
Comparison of the electrochemical profile of H2PD1-3 with that of natural prodigiosin revealed similar irreversible redox processes associated with several available redox events and ultimately leading to decomposition of the ligand. Notably, the peak potential for the first anodic event (Epa vs Fc/Fc+ in CH2Cl2) is tuned by the expected electronic effects given the different substitution patterns (i.e., alpha-ester vs alpha-t-Bu, meso-phenyl vs meso-H). For instance, consistent with the more electron-withdrawing nature of the ester group as compared to the tert-butyl group, Epa1 for H2PD1 is higher than that for H2PD2 by approximately 400 mV. These comparative data document the electronic tunability of conjugated pi-systems in pyrrolyldipyrrins.
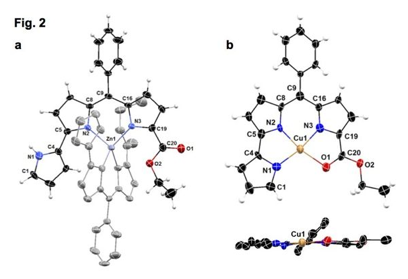
Our first investigation of the coordination ability of these new tripyrrolic ligands focused on H2PD1, in which the alpha-ester group was envisioned as an additional neutral coordinating group. In the presence of Zn(II) ions in THF or methanol, H2PD1 behaves as a bidentate monoanionic ligand (Fig. 2a).4 Conversely, this ligand system binds Cu(II) ions with 1:1 stoichiometry as a tetradentate dianion and leads to pseudo-square planar complex Cu(PD1) (Fig. 2b). Unlike previously reported studies of copper binding in pyrrolyldipyrrins,2,4 this insertion did not require the addition of a base and was not affected by complications arising from interfering redox events. The coordination environment of the copper center in Cu(PD1) was investigated in solution by electron paramagnetic resonance (EPR) spectroscopy. The 14N hyperfine splittings in the continuous-wave EPR spectrum were not sufficiently resolved for the determination of the number of copper-bound nitrogen atoms, therefore we employed pulsed electron-nuclear double-resonance technique ENDOR and identified three different nitrogen donors with different hyperfine interaction constants, thus demonstrating the solution structure of the complex.
Cyclic voltammetry data for Cu(PD1) indicate that multiple irreversible redox processes for this complex are associated with decomposition/demetalation pathways. In order to investigate the redox properties of metal-bound PD12-, we therefore sought to coordinate a more inert ion that would present a similar square planar geometry. The palladium(II) complex Pd(PD1) was isolated and its cyclic voltammogram displays a reversible one-electron reduction at E1/2 = -1.35 V with Epa/Epc separation of 68 mV. A cathodic event at similar potential (approximately -1.25 V) is observed for Cu(PD1), but it is irreversible in that case. Our DFT calculations indicate that these reductive processes are primarily ligand-based, leading to the population of molecular orbitals of antibonding nature with little (if any) metal-based character. These experiments demonstrated that metal-bound PD12- offers accessible one-electron ligand-based pathways of reactivity and the ligand-based reduction of Pd(PD1) could lead to a stable coordinated radical on the tripyrrolic ligand.
Part of our work on H2PD1 has recently appeared in a full paper on Inorganic Chemistry. Investigations on the metal binding properties of the other prodigiosins in our series, as well as the redox behavior of the coordinated scaffolds, are underway. Concurrently, we are exploring the oxidative chemistry of the isolated complexes both in aqueous solutions and in organic solvents. Funding for this project has allowed for the training of one graduate student, one undergraduate student and one postdoctoral associate. These researchers have gained considerable experience in synthetic chemistry and in the characterization of metal complexes using multiple techniques, including UV-visible absorption, NMR, and EPR spectroscopies, X-ray diffraction and electrochemical methods.
Our findings show that the selected pyrrolyldipyrrins present diverse coordinating abilities and highly tunable redox properties. Supported by experimental and computational evidence, we demonstrated that the availability of high-lying ligand-based molecular orbitals leads to complexes that feature both metal-based and ligand-based redox processes, as well as potential for ligand-based radical stabilization. These characteristics are particularly advantageous for the engineering of reactivity in complexes of late transition metals, which present high d electron counts and multiple available oxidation states. The resulting redox systems will be examined in detail to build a framework for the design of contemporary catalysts for oxidative transformations including functionalization of C-H bonds in hydrocarbons.
(1) Furstner, A., Angew. Chem., Int. Ed. 2003, 42, 3582-3603.
(2) Regourd, J.; Ali, A. A.-S.; Thompson, A., J. Med. Chem. 2007, 50, 1528-1536.
(3) Diaz, R. I. S.; Bennett, S. M.; Thompson, A., ChemMedChem 2009, 4, 742-745.
(4) Park, G.; Tomlinson, J. T.; Melvin, M. S.; Wright, M. W.; Day, C. S.; Manderville, R. A., Org. Lett. 2003, 5, 113-116.










