Reports: DNI152183-DNI1: Sustainable Catalysts for Feedstock Chemical Functionalization
 printer friendly
printer friendlyUrea Activation of Diazo Compounds.
Building on our 2012-2013 ACS PRF-funded discoveries, further advances in metal-free insertion reactions of diazo compounds have been made. A breakthrough in the promise of urea-catalyzed activation of diazo compounds led us to discover an interesting, unprecedented N–H insertion/arylation reaction of nitrodiazo compounds in early 2012.1
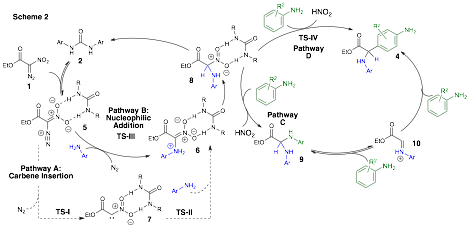
Investigations, both experimental and computational, probing the mechansim of our newly discovered urea-catalyzed multicomponent coupling reactions of nitrodiazoesters suggested the process was likely proceeding through a polar reaction pathway (Scheme 2). More specifically, the predominate reaction pathway likely involves urea activation of the diazo compound (5) for attack by aniline with concomitant extrusion of nitrogen gas to yield adduct 6. After proton transfer, intermediate 8 is presumed to form. A subsequent attack of 8 by a second equivalent of aniline followed by iminium ion (10) formation and finally a Friedal-Crafts reaction is proposed to complete the pathway in the formation of observed product 4. This work was carried out in collaboration with the Badjic and Hadad Research Groups and was published as a Feature Article in the Journal of Organic Chemistry in the summer of 2014.2

Enantioselective N–H Insertion/Arylations.
During the course of our mechanistic studies in the metal-free activation of 1, it was discovered that the newly formed stereogenic center in 4 can be controlled with a chiral phosphoric acid-derived catalyst (Scheme 3).3 Interestingly, the ability to control the enantioselectivity of these interesting multicomponent coupling processes is currently limited to chiral phosphonates: all of our attempts to control the stereochemical outcome with urea catalysts were unfruitful. Under the influence of optimal phosphoric acid derived catalysts, glycines 4 can be isolated in excellent yields with moderate control over the enantioselectivity.
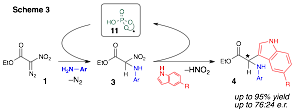
O–H and S–H Insertion Reactions.
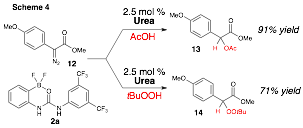
The scope of our urea-catalyzed insertion chemistry was extended to include aryldiazoacetates and a variety of O–H and S–H insertion partners (Scheme 4).4 For example, boronate urea 2a was found to be an excellent catalyst for insertion reactions of acetic acid enabling generating of 13 in excellent yield (91%). Similarly, urea-catalyzed insertion reactions of peroxides into aryl diazoacetates was developed; for example, 14 was formed in high yield (71%) under the influence of just 2.5 mol % of 2a.
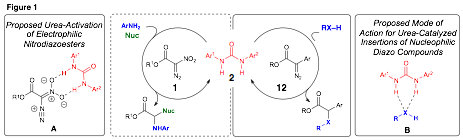
Mechanistic studies probing the role of 2a in reactions of aryldiazoacetates suggested that, contrary to our initial hypothesis, the urea is likely involved in amplifying the acidity of the insertion partner (B, Figure 1).5 The enhancement of the acidity of the organic acid enables the diazo compound 12 to deprotonate it and begin the catalytic pathway. This observation, in conjunction with our prior work with nitrodiazoesters, led to two unique modes of activation for ureas with respect to diazo insertion reactions: (1) ureas activate electrophilic diazo compounds for reaction with nucleophilic anilines via proposed complex A and (2) ureas catalyze insertion reactions of basic/nucleophilic diazo compounds through activation of the insertion partner.
Chiral Silanediols in Anion-Binding Catalysis.
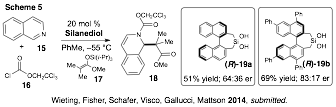
Since our discovery of enantioselective silanediol-based anion-binding catalysis 2013,6,7 our continued investigations of BINOL-based silanediols in N-acyl Mannich reactions of silyl ketene acetals led to an improvement in enantiomeric excess from the originally reported 20% ee to 80% ee. Several factors played critical roles in the enantiomeric excess enhancement, including: silanediol catalyst structure, silyl ketene acetal structure, and the reaction concentration (Scheme 5). From our optimization studies emerged a set of conditions that gave good yields of product with high levels of enantiocontrol. Specifically, the addition of TIPS-protected silyl ketene acetal 15 to the isoquinolinium ion generated in situ from 15 and 16 gave rise to the desired product in 69% yield and 83:17 er under the influence of 4,4',6,6'-substituted tetraphenyl catalyst (R)-19b. In contrast, the unsubstituted BINOL-derived silanediol (R)-19a resulted in both lower yields and lower enantioselectivity for the same reaction.
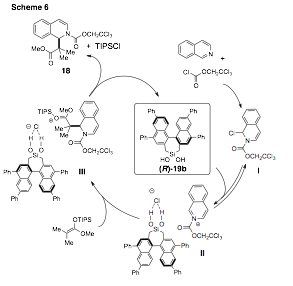
 As part of program focused on understanding the role of the silanediol catalyst, the acidities and binding constants were studied for select BINOL-based silanediols. The silanediols were significantly less acidic than anticipated: both silanediols (R)-19a and (R)-19b had a pKa(DMSO) near 19. The binding constant with tetrabutylammonium chloride in CDCl3 for (R)-19a and (R)-19b were 219 M-1 and 310 M-1, respectively. The data collected in these studies, combined with results from the literature, enabled us to propose an anion-binding mechanism for the silanediol-catalyzed addition of silyl ketene acetals to N-acyl isoquinolinium ions (Scheme 6). It is proposed that the silanediol facilitates ionization of I to generate ion pair II. Stereoselective addition of 17 to II then gives rise to III. The observed product 18 is formed with the simultaneous formation of TIPSCl and (R)-19b.
As part of program focused on understanding the role of the silanediol catalyst, the acidities and binding constants were studied for select BINOL-based silanediols. The silanediols were significantly less acidic than anticipated: both silanediols (R)-19a and (R)-19b had a pKa(DMSO) near 19. The binding constant with tetrabutylammonium chloride in CDCl3 for (R)-19a and (R)-19b were 219 M-1 and 310 M-1, respectively. The data collected in these studies, combined with results from the literature, enabled us to propose an anion-binding mechanism for the silanediol-catalyzed addition of silyl ketene acetals to N-acyl isoquinolinium ions (Scheme 6). It is proposed that the silanediol facilitates ionization of I to generate ion pair II. Stereoselective addition of 17 to II then gives rise to III. The observed product 18 is formed with the simultaneous formation of TIPSCl and (R)-19b.
Select References.
1. So, S. S.; Mattson, A. E. J. Am. Chem. Soc. 2012, 147, 8798. 2. So, S. S.; Oottikkal, S.; Badjic, J. D.; Hadad, C. M.; Mattson, A. E. J. Org. Chem. 2014, 79, 4832. 3. So, S.; Mattson, A. E. Asian J. Org. Chem. 2014, 3, 425. 4. Couch, E. D.; Auvil, T. J.; Mattson, A. E. Chem. Eur. J. 2014, 20, 8283. 5. For recent examples of proposed urea/Br¿nsted acid co-catalysis, see: a) N. Mittal, D. X. Sun, D. Seidel, Org. Lett. 2014, 16, 1012–1015; b) C. Min, N. Mittal, D. X. Sun, D. Seidel, Angew. Chem. Int. Ed. 2013, 52, 14084–14088; c) N. Z. Burns, M. R. Witten, E. N. Jacobsen, J. Am. Chem. Soc. 2011, 133, 14578–14581; d) H. Xu, S. J. Zuend, M. G. Woll, Y. Tao, E. N. Jacobsen, Science 2010, 327, 986–990; e) R. S. Klausen, E. N. Jacobsen, Org. Lett. 2009, 11, 887–890. 6. Schafer, A. G.; Wieting, J. M.; Fisher, T. J.; Mattson, A. E. Angew. Chem. Int. Ed. 2013, 52, 11321. 7. For a recent review on anion-binding catalysis, see: Brak, K.; Jacobsen, E. N. Angew Chem. Int. Ed. 2013, 52, 603.










