Reports: ND653263-ND6: Characterization of Nanodiamonds in Complex Environments
 printer friendly
printer friendlyThis Progress Report addresses four research directions:
1. Integration of GAMESS with QMC programs:
To facilitate the exchange of data between GAMESS [1] and other computational science packages, the long-term support (LTS) GAMESS standard output format (*.gamess) was developed.
The LTS .gamess format is used for the integration of GAMESS with the QMC program QWalk [2].
2. QMC integrated with the effective fragment molecular orbital (EFMO) method:
The EFMO method [3] combines the Fragment Molecular Orbital (FMO) and Effective Fragment Potential (EFP) methods [4].
In the newly developed QMC-EFMO method, the energy up to dimer corrections is given by
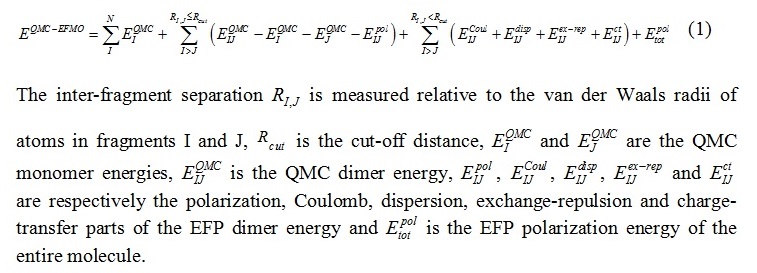
The QMC-EFMO method was initially tested on a four-water system (see Fig. 1).
Fig. 1: The four-water structure (taken from the Cambridge Cluster Database [5]).
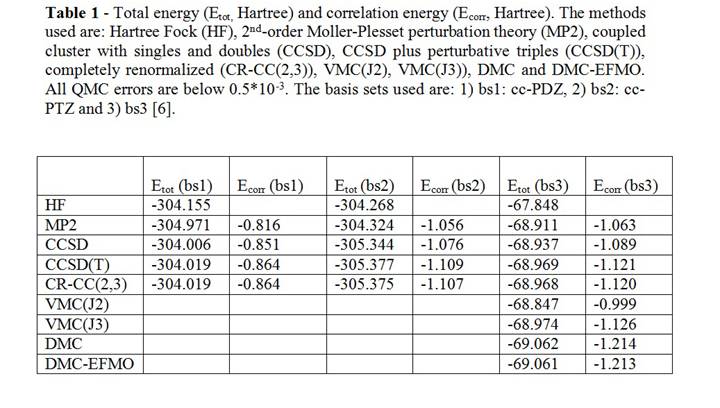
Table I presents the correlation energy of the four-water system by different high-quality quantum chemical methods including Variational Monte Carlo (VMC) with optimized two-body (J2) and three-body (J3) Jastrow terms, Diffusion Monte Carlo (DMC) and DMC-EFMO. The results demonstrate that DMC recovers more correlation energy than CCSD(T) and CR-CC(2,3) and that DMC-EFMO recovers almost entirely the DMC correlation energy.
The QMC-EFMO implementation is currently tested on water clusters of different sizes. Linear scaling of the QMC-EFMO computational cost with the system size is expected. The QMC-EFMO error will grow only linearly with the system size.
A journal article is in preparation.
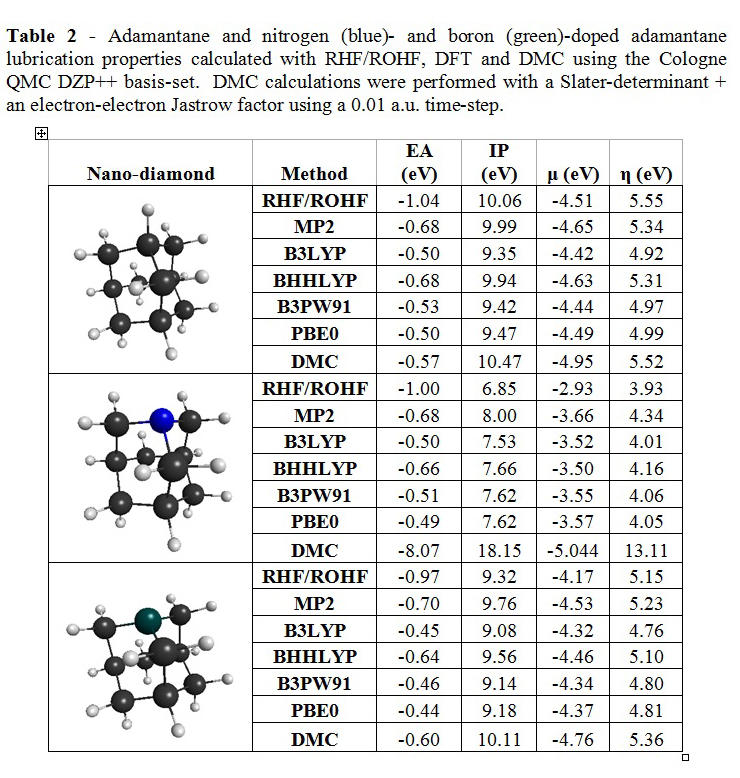
3. Engineering the lubrication properties of nanodiamonds with doping:
Chemical potential and chemical hardness (Equations 2a and 2b, respectively) are the primary molecular properties used to predict a material’s ability to lubricate. The chemical potential and hardness of a molecular lubricant are approximated in terms of the ionization potential (IP) and electron affinity (EA) [7].
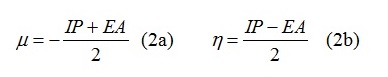
DFT, MP2 and DMC (QWALK) were applied to study the effect of n- and p-type doping of adamantine on the chemical potential and hardness. DMC values for adamantane agree with recent literature [8]. Both n- and p- doping result in reduced chemical potential and hardness, although only slightly for n-doping.
4. QMC forces with Algorithmic Differentiation (AD):
A prototype VMC-AD program was developed, which computes the interatomic VMC force in the ion by AD. The results by the prototype program are shown in Fig. 2. The forces are computed (1) by AD using the Hellman-Feynman and Pulay terms, (2) by AD using only the Hellman-Feynman term and (3) by correlated-sampling finite differencing for comparison [9, 10]. The Rapsodia AD computational tool [11] was used. The agreement between the AD (using Hellman-Feynman and Pulay terms) and finite-differencing forces confirm the correctness of the prototype.
References:
1. M.W.Schmidt, K.K.Baldridge, J.A.Boatz, S.T.Elbert, M.S.Gordon, J.H.Jensen, S.Koseki, N.Matsunaga, K.A.Nguyen, S.Su, T.L.Windus, M.Dupuis, J.A.Montgomery, J. Comput. Chem., 14, 1347-1363(1993); M.S.Gordon, M.W.Schmidt pp. 1167-1189, in "Theory and Applications of Computational Chemistry: the first forty years" C.E.Dykstra, G.Frenking, K.S.Kim, G.E.Scuseria (editors), Elsevier, Amsterdam, 2005.
2. L. K. Wagner, M. Bajdich, and L. Mitas, J. Comp. Phys. 228, 3390 (2009).
3. S. R. Pruitt, C. Steinmann, J. H. Jensen, and M. S. Gordon, J. Chem. Theory Comput. 9, 2235 (2013).
4. M. S. Gordon, J. M. Mullin, S. R. Pruitt, L. B. Roskop, L. V. Slipchenko, and J. A. Boatz, J. Phys. Chem. B 113, 9646 (2009).
5. The Cambridge Cluster Database, D. J. Wales, J. P. K. Doye, A. Dullweber, M. P. Hodges, F. Y. Naumkin F. Calvo, J. Hernández-Rojas and T. F. Middleton. (URL://www-wales.ch.cam.ac.uk/CCD.html)
6. M. Burkatzki, C. Filippi, M. Dolg, J. Chem. Phys. 126, 234105 (2007).
7. Shenghua, Li.; He, Yang.; Yuansheng, J. Int. J. Mol. Sci. 5, 13-34 (2004).
8. Drummond, N.D.; Williamson, A.J.; Needs, R.J.; Galli, G. PRL 95, 096801 (2005).
9. S. Sorella and L. Capriotti, J. Chem. Phys. 133, 234111 (2010).
10. M. Barborini, S. Sorella, and L. Guidoni, J. Chem. Theory Comput. 8, 1260 (2012).
11. Rapsodia: User Manual, I. Carpentier, J. Utke. (URL://www.mcs.anl.gov/Rapsodia/)










