Reports: ND452655-ND4: Hydrogen Bond Catalysis and Anion Molecular Recognition
 printer friendly
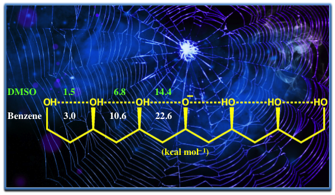
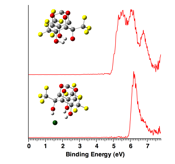
printer friendlyThe strengths of different types of hydrogen bonds in
hydrogen bond networks (HBNs) were quantified in the gas phase and in solution
via photoelectron spectroscopy and p
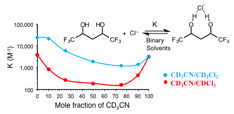
Anion binding constants of two flexible diols were
determined in different solvents and binary mixtures. The association constants
(
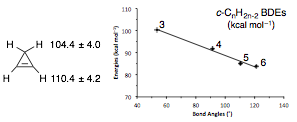
Work that had previously been carried out on reactive
intermediates with partial support from the Petroleum Research Fund was
published in