Reports: UR651152-UR6: A Computational Exploration of the Stereoselective Synthesis of Substituted Pyrrolidines
 printer friendly
printer friendlyThe goal of this funded research project is to investigate the stereospecific synthesis of substituted acyl pyrrolidines from a computational perspective in order to inform and guide the choice of experimental conditions leading to maximum stereoselectivity. The synthetic pathway under consideration is the tandem aza-Cope – Mannich reaction, an efficient method to the formation of a variety of substituted pyrrolidines that can serve as asymmetric organocatalysts.
During this third and final year of the grant we have focused on the following aspects of the reaction:
1. exploring an alternative mechanism for substrates with an electron-withdrawing substituent;
2. evaluation of a large chiral catalyst and its influence on stereoselectivity;
3. the role of substituents at the vinylic position on product selectivity.
For each of the substrates described below, we map the energy profile for the key steps of the reaction, including coordination of the acid catalyst to the oxazolidine starting material to produce the iminium cation intermediate, a series of C-C bond rotations that lead to the other possible stereoconformers and can interconvert among them, and the rate-determining aza-Cope rearrangement for each of the stereoisomers. A comparison of the activation barriers for these key steps will shed light on the factors that determine the observed stereochemical product ratios and enable us to suggest ways to improve it.
Results obtained to date are described below:
1. There is some evidence in the literature that conversion of an oxazolidine to the pyrrolidine product could proceed via an alternative mechanism, especially when the protecting group at the nitrogen center is electron withdrawing.[1] The alternative mechanism is also a tandem reaction: aza-Prins cyclization to a 6-membered intermediate followed by pinacol rearrangement to the pyrrolidine product (see Scheme below). Since the two mechanisms may give rise to significant differences in the stereochemical product ratio, we modeled the reaction using both mechanisms (aza-Cope – Mannich and aza-Prins – pinacol) and compared relative energy of intermediates and activation barriers. Specifically, we examined an electron-donating R1 protecting group (alpha-methylbenzyl) and an electron-withdrawing group (tosyl). We also compared substrates with a secondary or tertiary carbinol carbon (R2).
Scheme
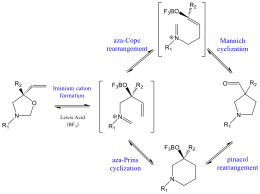
The results indicate that in the alpha-methylbenzyl system the aza-Cope pathway is thermodynamically favored over the aza-Prins pathway. However, when the protecting group is the electron-withdrawing tosyl group, the reaction still proceeds along the aza-Cope-Mannich pathway, but in a single concerted step. In other words, the rearranged aza-Cope intermediate is not a minimum on the potential energy surface along the intrinsic reaction coordinate. This is true for substrates with either a secondary or a tertiary carbinol carbon. These results indicate that the presence of the tosyl protecting group leads to significantly different reactivity patterns and it may result in improved stereoselective ratio of products.
2. The goal for this portion of the project was to determine whether a chiral Lewis acid could selectively react with one enantiomer of the oxazolidine ring starting material for a series of racemic starting materials in the initial ring-opening step of the reaction. This in turn would obviate the need to use enantiopure starting material. To this end, we compared two Lewis acids: the achiral boron trifluoride and the chiral isopinocampheyldifluoroborane. The chiral Lewis acid appears to show some selectivity for the α oxazolidine ring in the case where a benzyhydryl protecting group is used and R2 is a methyl group. However, when a tosylate protecting group is used, the chiral Lewis acid is selective for the α oxazolidine only if the group is oriented over the oxazolidine ring. In summary, there is some evidence that product selectivity might be improved by using a large chiral catalyst, a tosylate protecting group, and a tertiary carbinol carbon. However, the improvement in selectivity afforded by using a chiral catalyst is not sufficient to justify the added difficulties that are introduced in the experimental procedure.
3. Here we investigated the effect of placing a substituent at the vinyl position of the oxazolidine starting material. We are in the process of mapping out the energy profile for the key steps in the reaction for the substrate with a phenyl substituent at the vinyl position and comparing it to that for the substrate with no substituent. In both cases the protecting group at the nitrogen center is benzhydryl. In the course of this investigation we realized that we need a more robust methodology for finding transition states for the C-C bond rotations that lead to stereoisomer interconversion. Since our conclusions about stereoselectivity rest on a comparison of the relative activation barriers between interconversion and rearrangement, it is particularly important to obtain accurate values.
This project is in collaboration with one of my colleagues at Eastern Michigan University, Prof. Harriet Lindsay, who is carrying out the experimental work. The grant has allowed me to make significant progress on the project by giving me the opportunity to hire student researchers. Prof. Lindsay and I plan to submit a grant proposal to NSF during the 2015 calendar year to secure continued funding for this joint project.
In addition, financial support from this grant provided my students with the opportunity to present their results at the ACS Fall National Meeting in San Francisco in August. Participation at this event gave them confidence in their abilities to do research at a professional level and an opportunity to hear about areas of chemistry other than our own computational field.
The increased activity in my lab has also been an excellent recruiting tool and I currently have seven students working on three projects.
[1] a) Armstrong, A., and Shanahan, S.E. Org. Lett. 2005, 7, 1335-1338; b) Armstrong, A., Bhonoah, Y., and Shanahan, S.E J. Org. Chem. 2007, 72, 8019-8024.










