Reports: DNI651837-DNI6: Theoretical Considerations of Conjugated Polymer Self-Assembly
 printer friendly
printer friendlyThis year we focused on several issues:
1) Understand the role of solvation in changing single chain polymer structure and dynamics
2) Understand the role of side chain length in poly-3-alkylthiophene structures in benzene
3) Understand the role of point defects in polymer structure and behavior as it pertains to regio-irregular defects resulting from poor polymer synthesis issues.
The project is ongoing, and a preliminary report detailing the findings is below:
Simulation Details:
Molecular dynamics simulations are conducted using Gromacs 4.5.5. The OPLS all-atom forcefield with modifications for use with conjugated polymer systems was used as outlined in existing literature. Each system was initialized by steepest descent energy minimization followed by 1 ns of NVT simulation. Next, 4 ns of NPT simulation at 300 K allowed the box size to adjust to equilibrate the solvent density. For production runs, each system was simulated for 150 ns with a time step of 2 fs. Each system was run at 300, 450 and 600K, in order to probe the effect of temperature on polymer structure and interactions with solvent, with temperature held constant using the velocity-rescaling thermostat. Pressure-coupling in the initialization steps used the Berendsen barostat. We have performed 300K simulations on a wide range of solvents and polymer types.
Results and Discussion:
1) Polymer structure
Molecular simulations can also be utilized to investigate the solubility of polymer chains in different solvent environments. Snapshots of the polymer backbone from the AA-MD simulations were first coarse-grained into a linear polymer by averaging the coordinates of the sulfur atoms from monomers i-1, i, and i+1 so that the contribution of each to the average were respectively ¼, ½, and ¼. This procedure is functionally identical to previous work studying the mechanical properties of actin filaments from AA-MD simulations. The following equation is used to calculate persistence length (Lp):
![]() (3)
(3)
where Lc is the contour length and θ(Lc) is the angle between the tangent vector at Lc and the tangent vector at a reference point on the polymer chain. The persistence length was averaged from calculations starting from six equally spaced points along the polymer backbone spaced such that each calculation was independent. The Kuhn length (b) is twice as large as the persistence length (Lp) and the simulated Kuhn length of P3HT in benzene, toluene, chloroform and DCB were determined to be 6.7 ± 0.3 nm, 6.7 ± 0.4 nm, 6.4 ± 0.1 nm and 6.0 ± 0.1 nm, respectively. These values are in very good agreement with those previously reported by McCullough et al., The reported uncertainty is the standard deviation arising from averaging the Kuhn length from two unique simulations.
2) Polymer solvation and role in mediating polymer structure
Figure 1 shows the pairwise distribution function for the solvent carbon with respect to specific P3HT ring atoms. All solvents are found to pack more closely with the sulfur atom and the C2-carbon compared with the other thiophene carbon atoms. This is the result of steric hindrance for solvent packing around the C1-carbon and C4-carbon (due to polymerization) and the C3-carbon (due to the aliphatic hexyl side chain). Figure 2 shows that there is also preferential orientation of the solvent molecule. Toluene is more likely to pack around the P3HT backbone with the methyl group (electron donating group) closer to the polymer chain. On the other hand, DCB is more likely to pack around the P3HT backbone with the chlorines (electron withdrawing group) shielded from the polymer chain. The shielding of chlorine near the P3HT backbone is also present in chloroform (Supporting Information). These preferential interactions show a propensity for polar solvent molecules to pack with the partial positive charge (δ+) closest to the polymer chain. This specific solvent-polymer interaction could be expected given that P3HT is more likely to experience p-type (i.e., positive) doping.
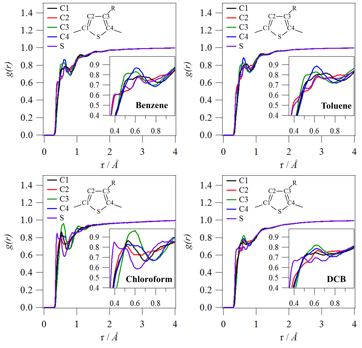
Figure 1. Pairwise distribution functions relating the specific sites along the P3HT backbone to the carbon atoms in the four different solvent molecules. In this plot, the specific backbone sites are decomposed into different curves, whereas the sites in the solvent molecules are averaged out.
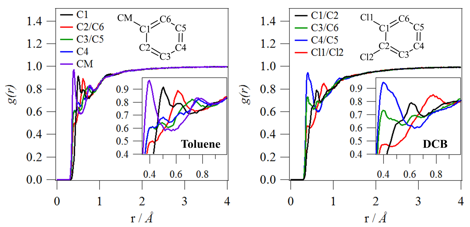
Figure 2.Pairwise distribution functions relating the carbon atoms along the P3HT backbone to specific atoms in a toluene molecule (left) and a DCB molecule (right). In this plot, the specific solvent atom sites are decomposed into different curves.
3) Point defects / regio-irregularities
Extensive simulations of 9 and 15- length P3HT polymers with various region-irregularities were performed in the solvents benzene and chlorobenzene. Figure 3 shows the types of defects that were considered and Figure 4 shows the probability distributions of the backbone torsional angles. The main conlusion is the point defects in the regio-regularity do not significantly affect the polymer conformation beyond the +1/-1 position.
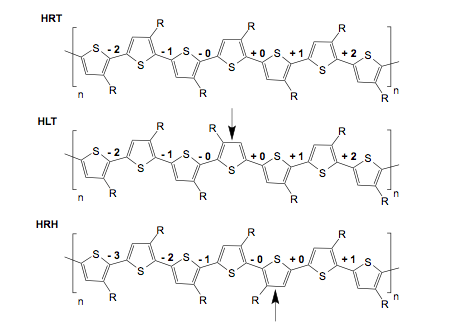
Figure 3: Schematic drawing of n=9 and n=15 mer simulations showing the region-irregularities at the +/- 0 positions
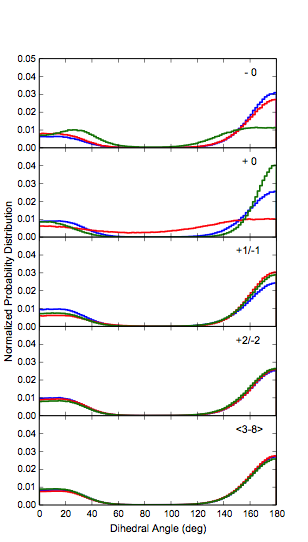
Figure 4: Normalized probability distributions for the polymer torsional backbones.










