Reports: DNI1052404-DNI10: Validating Computational Design Principles for Crystalline Enzyme Assemblies
 printer friendly
printer friendlyOverview. New design principles and multi-state design algorithms are required to build new crystalline assemblies from protein building blocks. In the first year of this work, we have made significant progress. Experimentally, our work has focused on the prerequisite step of identifying suitable building block proteins. Computationally, we have successfully created an efficient algorithm for "searching crystal space" to identify crystalline assemblies that are designable and have other topological properties of interest. We are now poised to design, express, and crystallize new assemblies in the second year of support.
Impact. The ACS PRF support has been extraordinarily helpful as a new investigator to tackle a project that is significantly ambitious. While control over protein assembly using computational design is a "hot area"1,2,3, a new laboratory nonetheless requires significant preliminary results to obtain funding. In addition to supporting a Ph.D. student, ACS PRF funding has gone for the training of multiple undergraduate students who have learned how to transform E. coli, express proteins, run denaturing protein electrophoresis experiments, purify proteins, and setup crystallization trials. These students have worked during the term and during the summer, learning foundational skills that are broadly applicable in future molecular biology settings.
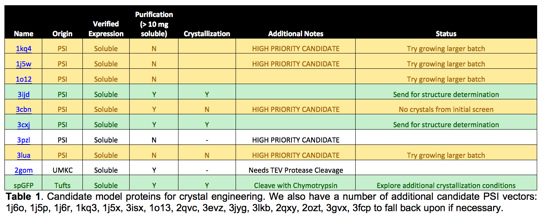
Experimental Progress. Ultimately, we aim to provide design algorithms capable of predicting surface mutations to convert arbitrary protein monomers into crystal building blocks. To develop such methods the first step is to identify a suitable model protein for demonstration and testing. Given that we may need to experimentally test multiple variants, the original model protein should be stable (to remain folded upon mutation), easy to purify and crystallize (for speed), and easy to express in E. coli. The most traditional model protein for crystallography, hen egg white lysozyme (HEWL) fails this last test; it is not easy to produce HEWL mutants in E. coli. To rapidly identify a replacement model protein we pursued a parallel strategy. Namely, we obtained 20 thermophilic proteins from the Protein Structure Initiative: Biology Materials repository, and have run a number of these targets through expression, purification, and crystallization trials (Table 1) to identify the best behaved protein to serve as a building block for building new crystal forms via design.
Computational Progress. To better understand the process of crystallization, and to design new protein crystals, it is helpful to generate a set of candidate crystal forms other than those observed experimentally. However, it is challenging to efficiently search the space of possible protein crystal structures. In addition to the varying symmetry requirements of the 65 permissible chiral space groups, there are multiple rotational and translational degrees of freedom. We have therefore developed an algorithm for sampling candidate protein crystals inspired in part by the Fast Fourier Transform (FFT) protein docking algorithm of Kaltchalski-Katzir4. Using a GTX Titan graphics card (GPU), CUDA, PyCUDA, and PyFFT, it is possible to score an array of discrete transformations very rapidly. Use of the GPU accelerates the FFT calculation by approximately a factor of 4-7 for cubic grids of varying size (64, 128, or 256). The resulting translational score arrays can be used to construct a greedy depth first search over possible Bravais lattice parameters. For example, in the P1 space group, the search is limited to the selection of three compatible unit cell axes. By carefully compositing the translational scoring array, and by pruning less promising edges from the tree it is possible to efficiently sample the most densely packed crystal forms. The new software for Python based protein docking will be included in the upcoming release update of SHARPEN, an open-source library for protein modeling and design5.
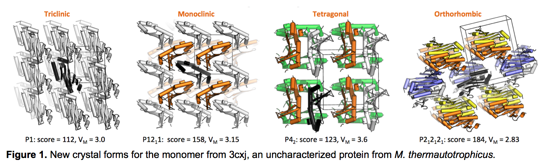
In addition to the first designed protein crystal from Saven and coworkers1 other recent milestones were the design of tetrahedral and octahedral oligomers from Yeates, Baker, and coworkers2,3. These studies began with a loosely specified packing arrangement, and search for the most promising assembly by varying several degrees of freedom within defined boundaries. The algorithm described here seeks to identify desirable packing arrangements without a preordained space group. Presumably, packing arrangements with more extensive interfaces will be more designable. 3cxj, an uncharacterized protein from M. thermautotrophicus, natively crystallizes in the P4322 space group. Our new algorithm generates new, densely packed, alternative crystal candidates (Figure 1). Moving forward, we will apply computational protein design methods to identify which alternative crystal packing is the most favorable for our candidate model proteins. We will select a limited number of designed variants for downstream validation. Validation will consist of expression, purification, crystallize, and x-ray crystallography structure determination to assess the design accuracy.
1. Lanci, C. J. et al. Computational design of a protein crystal. Proc. Natl. Acad. Sci. U. S. A. 109, 7304–7309 (2012).
2. Lai, Y.-T., Cascio, D. & Yeates, T. O. Structure of a 16-nm Cage Designed by Using Protein Oligomers. Science 336, 1129–1129 (2012).
3. King, N. P. et al. Computational Design of Self-Assembling Protein Nanomaterials with Atomic Level Accuracy. Science 336, 1171–1174 (2012).
4. Katchalski-Katzir, E. et al. Molecular surface recognition: determination of geometric fit between proteins and their ligands by correlation techniques. Proc. Natl. Acad. Sci. U. S. A. 89, 2195–2199 (1992).
5. Loksha, I. V., Maiolo, J. R., 3rd, Hong, C. W., Ng, A. & Snow, C. D. SHARPEN-systematic hierarchical algorithms for rotamers and proteins on an extended network. J. Comput. Chem. 30, 999–1005 (2009).









