Reports: ND451880-ND4: The Selective and Sequential Chemistry of Electronically-Coupled Lactones
 printer friendly
printer friendlyBackground and significance.
Synthetic methodology to rapidly and efficiently prepare multifunctional molecules continues to leverage discoveries in disciplines spanning materials science and chemical biology. One approach to discrete multifunctional molecules is illustrated in Figure 1. A statistical mixture (with the percentages shown) results if the reaction rates between reagent site B and positions A, A′, and A″ are identical (top pathway). If each successive functionalization reaction sufficiently deactivates the remaining reactive sites, and a reactivity gradient is established, a one-pot sequential multifunctionalization can be executed (bottom pathway).
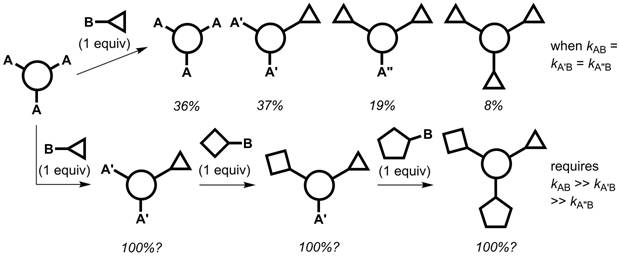
Figure 1. The kinetics associated with statistical and non-statistical syntheses of discrete multifunctional molecules from a three-fold symmetrical precursor.
We recently reported that benzotrifuranone (BTF) 1 (Scheme 1) is uniquely suited for multifunctionalization through sequential aminolysis reactions.1 The platform affords rapid access to mono- (2), di- (3), and trifunctionalized (4) targets provided routine control of temperature and amine reagent stoichiometry, and lends itself to the one-pot synthesis of multifunctionalized 4 (i.e., 4R1R2R3) from 1 in excellent yield (85%) and a single day. We initially proposed an electronic/inductive argument to rationalize the kinetic deactivation observed upon successive aminolysis of 1. In this funding period we have obtained a more comprehensive understanding of the aminolysis behavior of BTF through comparative kinetics measurements, X-ray crystal structure analysis, and quantum chemical calculations
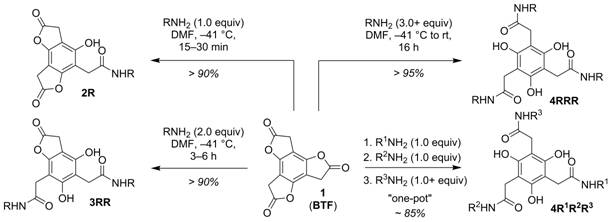
Scheme 1. Aminolysis of benzotrifuranone (BTF) 1.
Aminolysis of benzotrifuranone (BTF): General features.
The ring opening of 1 proceeds efficiently and in a stepwise fashion (i.e., 1→2→3→4, Scheme 1) in the presence of primary and cyclic secondary amines, including amines with a-branching, chiral centers (e.g., amino acids), and those introduced as their salts (in the presence of an exogenous base). The lactone reactivity gradient established by 1, shown earlier,1 makes possible the one-pot preparation of trifunctionalized 4 using three different amines.
Aminolysis of benzotrifuranone (BTF): Kinetics.
Rate data for the stepwise aminolysis of 1 were obtained and have set the stage for better understanding the system. The results for the n-heptylaminolysis of 1 at 24 °C in acetonitrile (performed under pseudo-first-order conditions) are shown in Figure 2 as measured by stopped-flow IR spectroscopy (by monitoring the appropriate lactone C=O resonance). The initial ring opening of 1 (t1/2 = 0.98 s) is rapid, about an order of magnitude faster than the aminolysis of p-nitrophenyl acetate, a conventional activated ester. The ring openings of 2a and 3aa are more than an order of magnitude slower and half an order of magnitude apart (with half-lives of 54 seconds and 540 seconds, respectively).
Structure and reactivity comparison of BTF to related benzoate esters.
The aminolysis behavior of four related benzoate esters (5–8, Figure 3) has been evaluated during this reporting period to shed light on the structural features responsible for the stepwise deactivation of 1. Included is ring-expanded benzotripyranone 5 (BTP), which could be prepared in one step through acid-catalyzed intramolecular transesterification of previously reported triisopropyl 3,3',3''-(2,4,6-trihydroxybenzene-1,3,5-triyl)tripropionate.2 Like BTF 1, BTP 5 forms a dilactone (not shown) as the major product upon reaction with one equivalent of aliphatic amines a–e. The reactions, however, under conditions identical to BTF aminolysis (–41°C in DMF), are considerably slower and lower yielding. These observations are consistent with the absolute aminolysis rates (Figure 3); the rate differences within the BTP (5) series are much smaller than those for BTF (1), leading to decreased aminolysis selectivity. Comparison of 1 and 5 with 6–8 is also insightful. Monolactones 7 and 3aa have very similar aminolysis rates, while 8 reacts slightly more slowly than the monolactone derived from 5. The acyclic and unstrained phloroglucinol triacetate (6) is significantly slower than all other compounds investigated.
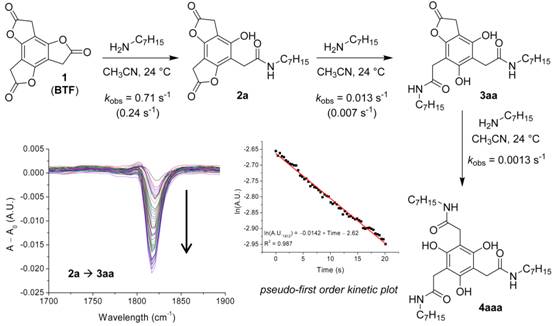
Figure 2. Kinetics of the n-heptylaminolysis of 1. The values shown in the scheme are averaged pseudo-first-order rate constants from multiple runs (amine concentration = 100 mM); statistically corrected values are shown in parentheses (values assume that the lactones of 2a react at similar rates). A representative kinetics run is shown.
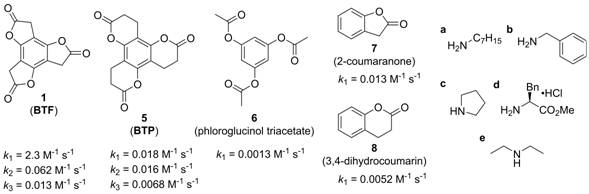
Figure 3. Additional benzoate esters investigated during this reporting period. Statistically-corrected second-order rate constants (calculated as kobs/[amine]) are shown for n-heptylaminolysis at 24 °C in acetonitrile.
Mechanistic and structural conclusions.
In the absence of kinetics data and comparison with 5–8, the stepwise kinetic deactivation of 1 upon aminolysis was previously attributed to electronic effects.1 Analysis of the kinetics data shown in Figure 3, as well as additional computational and X-ray crystallography data, now point more strongly to a “ring strain gradient” as the most significant selectivity driver for 1. Take, for example, the aminolysis of 6 which is seven times faster (on a per ester basis) than simple phenyl acetate (based on data available from the literature). A similar (four-fold) reactivity difference is found between the k1 values of 5 and 8, consistent with an electronic argument that considers substituent effects involving the central aromatic ring. The much larger 180-fold difference between the k1 values of 1 and 7 speaks to a significant change in structure, and overall ring strain. Sequential ring strain release, coupled with a favorable electronic gradient, appears to be the basis for the unusual stepwise aminolysis behavior of 1. Speaking to the strain associated with 1 (relative, for example, to 5) is bond length alternation within its benzene core (1: ±0.021 Å; 5: ±0.003 Å) based on X-ray crystal structure analysis.
Ongoing work.
The work described above, completed primarily by one graduate student, will be the basis for a soon submitted full paper.3 Ongoing work, being performed by two other graduate students, seeks to a) develop a novel, efficient synthesis of 1 and b) utilize 1 in the preparation of functional molecular systems and materials.
References.
1. Baker, M. B.; Ghiviriga, I.; Castellano, R. K. Chem. Sci. 2012, 3, 1095–1099.
2. Lampkins, A. J.; Li, Y.; Al Abbas, A.; Abboud, K. A.; Ghiviriga, I.; Castellano, R. K. Chem. Eur. J. 2008, 14, 1452–1463.
3. Baker, M. B.; Tasseroul, J.; Lampkins, A. J.; Al Abbas, A.; Abboud, K. A.; Ghiviriga, I.; Castellano, R. K., manuscript in preparation.









