Reports: DNI1050934-DNI10: Effect of Surface-to-Bulk Atomic Structure on Surface Electronic Structure in Multicomponent Catalysts
 printer friendly
printer friendlyThe motivation for our ACS-PRF project came from our earlier work where we had set out to explore the fundamental structure-property relationships of Pt-TiO2 hybrid surfaces and demonstrating the application of these materials for methanol electro-oxidation. Specifically, we investigated the relationship between the atomic and electronic structure of Pt to the activity of the resulting Pt-TiO2 towards methanol oxidation [ACS Appl. Mat. & Interfaces, Vol. 3, 2, 147-151, 2011]. What we had found was that the platinization of highly ordered, vertically oriented titania nanotube arrays (TNT) through a Cu precursor could be used to control atomic-electronic structure of the surface Pt and, as a result, could be used to tune its catalytic activity. We found that increasing the metal-support distance, by increasing the thickness of the Cu intermediary layer, the surface Pt is rendered metallic Pt rather than cationic, which dramatically increases the activity of Pt for methanol electro-oxidation.
The current ACS-PRF grant has allowed us to begin more in-depth investigations of the near-surface structure-property relationships in catalysts. The near-surface compositional architecture in photo, electro, and traditional heterogeneous catalyst systems fundamentally shapes their properties. Design of near-surface composition often employs synthesis through solution chemistry where metal ions are reduced onto a support surface. It is this area of solute-derived catalyst metal deposits that we initially focused our attention on. It is nominally understood that as each layer of metal deposits build, the reduction reaction is complete and that the properties of the adlayer metal may be described by those of neutrally charged atoms. There is also an expected threshold lengthscale over which the support material affects the surface chemistry through electron transfers. However, for very low adlayer thicknesses of deposited species, it is not clear whether the deposit prefers to remain partially charged through effects of either low dimensionality or charge transfer with the substrate.
What we found, through core-electron spectroscopy and cyclic voltammetry combined with corroborating DFT calculations, is that submonolayer, single monolayer, bi- and tri-layer Pt architectures on Au display unique properties arising from low-dimensionality and electron transfer phenomena. Solute derived submonolayer Pt has a strong preference for remaining cationic rather than neutral on a Au surface and consequently exhibit deviated catalytic activity compared to bulk Pt. This study has revealed that the nature of ultrathin Pt films undergoes a phase transition below a threshold distance of 2-3 monolayers from a Au surface, in terms of its atomic/electronic structure and
| Figure 1: Schematic of the Pt-Au surface for three different thicknesses of Pt, indicating the prevalence of cation-anion complexes for thinner Pt deposits. Reaction arrows (orange) indicate CO electro-oxidation (left) and film dissolution (right). |
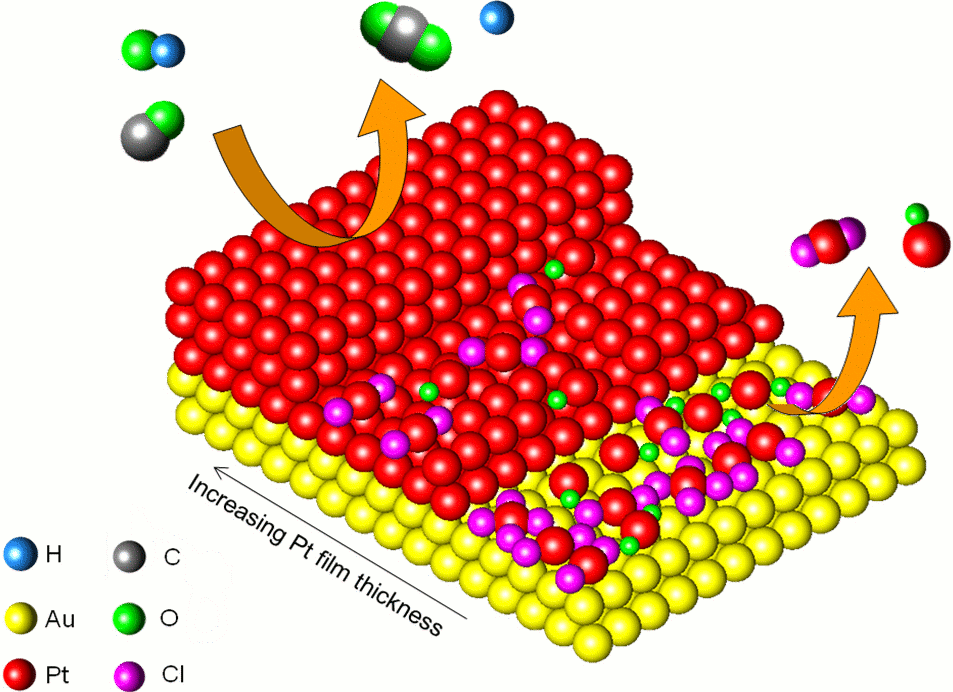
the resulting chemical properties, such as electrochemical activity for methanol oxidation (Figure 1 and 2). Based on these monolayer catalysts, we created a hybrid structures using Au/CFP (Au on carbon fiber paper) as promising electrode architectures for direct methanol fuel cells (DMFCs), and investigated the fundamental relationship between the near-surface Pt atomic/electronic structure and the activity of this hybrid electrocatalyst towards methanol oxidation. Synthesized Au/CFP heterostructures were found to depend on the type of Au electrodeposition method employed.
| Fig.2: The durability of Pt electrocatalysts is shown as a function of the Pt layer thickness. The initial activity, the rate of change of the activity with electrochemical cycling and the final steady-state activity all depend on the Pt monolayer thickness. |
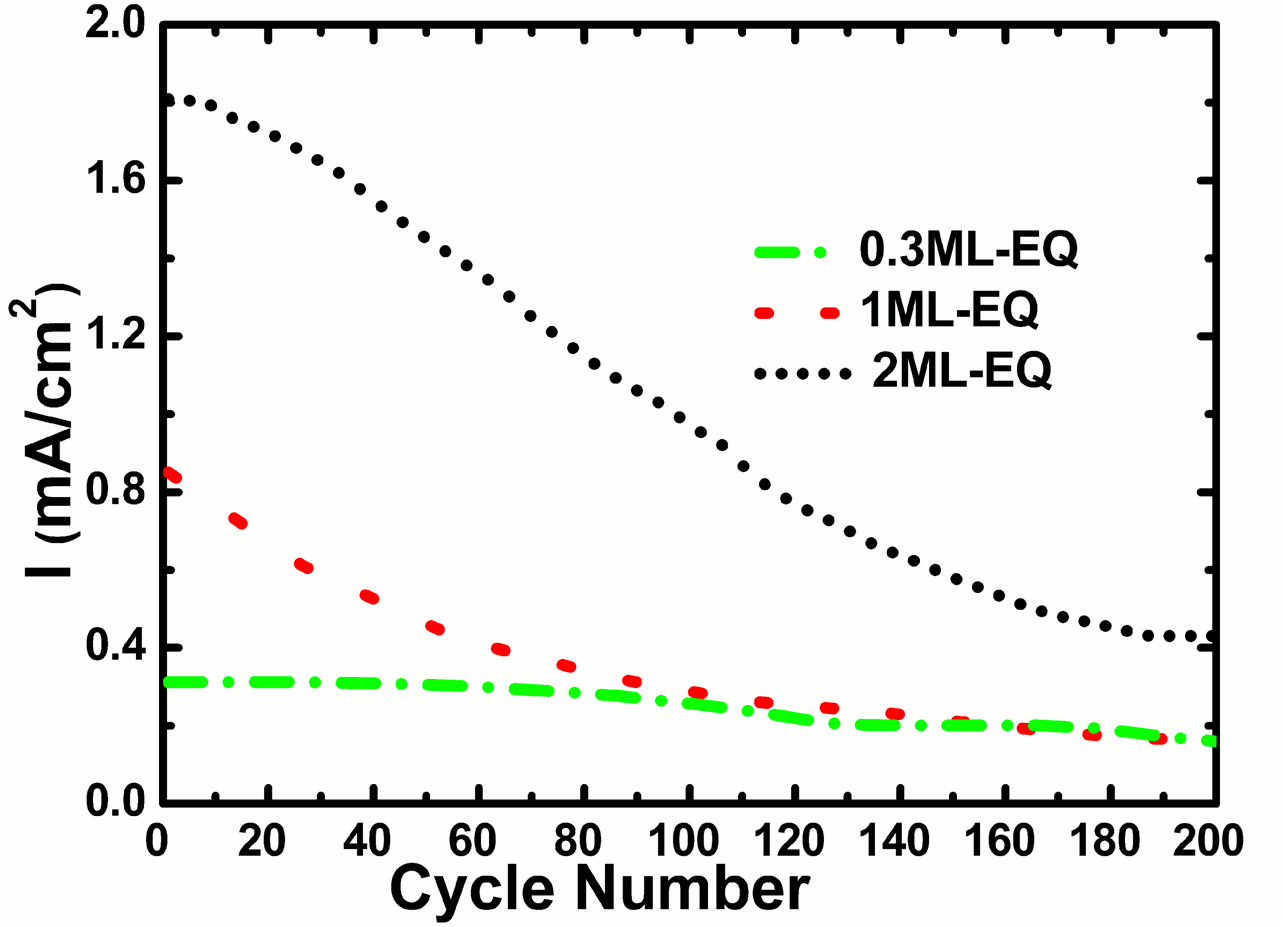
The properties of the Pt near-monolayer deposits were found to depend strongly on the morphology of the support with varying propensity for a cationic chemical state. A survey of literature shows that this is one of the first reports where the scientific relationship between the electrochemical activity and structural durability of ultrathin Pt films, has been explicitly related to the fundamental measurements of the evolution of atomic and electronic structure during layer by layer Pt growth. This study shows that an equivalent of two monolayers (ML) is the low-loading limit of Pt on Au. At one ML or below, the Pt film decreases in activity and durability very rapidly due to presence of cationic Pt. This finding is of critical importance in the design of low-dimensional corrosion tolerant Pt catalysts in nanoparticle, thin-film or core-shell geometries. [ACS Appl. Mat. Interf., Vol. 3, 10, p. 3948-3956, 2011]
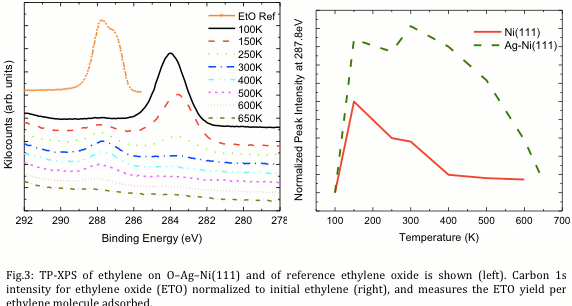
In a simulytaneous study, we explored the surface tunability of gas-phase catalytic activity through designed near-surface architecture using the epoxidation reaction as a model. Epoxidation is an industrially ubiquitous process in the synthesis of epoxy, which is used in adhesives, surface coating materials, and structural materials. In this work it was discovered through a combination of low energy electron microscopy (LEEM) and temperature programmed X-ray photoelectron spectroscopy (TP-XPS), that terrace- and step-edge-grown Ag on Ni(111) that the addition of submonolayer Ag to a Ni(111) surface increases the activity of the surface for ethylene epoxidation (Figure 3) while simultaneously stabilizing the ethylene oxide produced from further reaction at elevated temperatures. By combining submonolayer amounts of Ag with Ni, two active metals were paired in a manner that retained their desired epoxidation and dehydrogenation activities while suppressing unwanted C–C bond breakage. This work shows the activity for epoxidation is significantly increased through tailored combinations of two different catalyst metals [PCCP, Vol. 13, 23, p.11034-11044, 2011]. More broadly, the methods in this study serve as a template for the catalysis community to follow.
| Fig.3: TP-XPS of ethylene on O–Ag–Ni(111) and of reference ethylene oxide is shown (left). Carbon 1s intensity for ethylene oxide (ETO) normalized to initial ethylene (right), and measures the ETO yield per ethylene molecule adsorbed. |