Reports: ND149109-ND1: Alkali and Alkaline Earth Metal Complexes for Catalytic Hydroaminations
 printer friendly
printer friendlyThe reaction mechanism of alkali, alkaline earth, and rare earth metal-based hydroamination catalysts is believed to proceed via the insertion of the C=C double bond into a metal-amide bond, followed by protonolysis to regenerate the catalytic metal amide species (Scheme 1). Rare earth metal-based catalyst systems are commonly thought to involve a rate-determining olefin insertion step and fast protonolysis. The relative rate of the two steps is less clear for catalysts based on alkaline earth metals, because these catalysts predominantly display a first order rate dependence on substrate concentration in disagreement to a fast protonolysis step. One hypothesis has been that a coordinated amine molecule could assist in a concerted non-insertive N-C ring closure with concurrent amino proton transfer from the amine onto the olefin (Scheme 2), effectively combining the insertion and protonolysis step to a single step. An alternative scenario would imply a reversible olefin insertion step and a rate-determining protonolysis step (Scheme 3). In order to probe these mechanistic scenarios we set out to study the mechanism experimentally and computationally in collaboration with Dr. Sven Tobisch at the University of St Andrews utilizing newly developed model catalyst systems prepared in our group (Scheme 4).
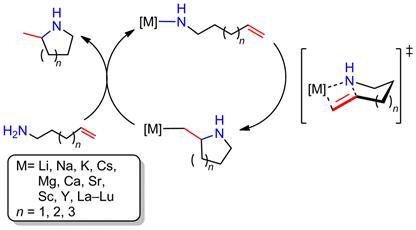
Scheme 1. Insertion mechanism for the hydroamination/cyclization of aminoalkenes.
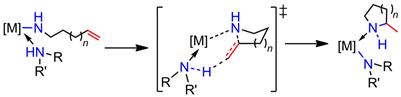
Scheme 2. Proposal for a concerted non-insertive N-C ring closure with concurrent amino proton transfer onto the olefin in the hydroamination/cyclization (RRxNH = substrate or hydroamination product).
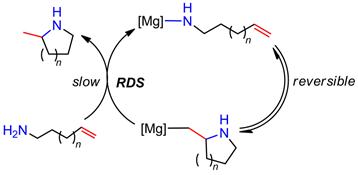
Scheme 3. Proposal for an alternative mechanistic scenario with reversible olefin insertion and rate-determining protonolysis (RDS = rate determining step).
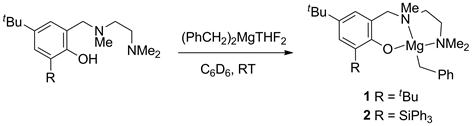
Scheme 4. Synthesis of phenoxyamine magnesium model catalyst systems.
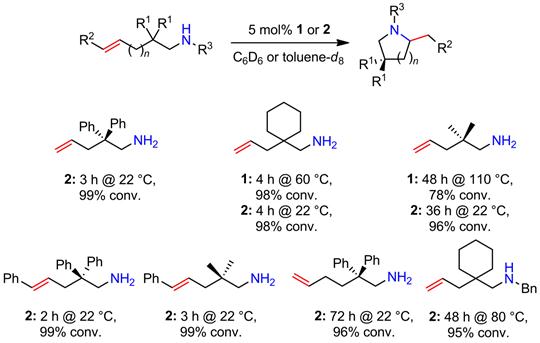
Figure 1. Catalytic hydroamination/cyclization of aminoalkenes using the model catalyst systems 1 and 2.
Complex 1 proved to be significantly less stable and also less catalytically active (Figure 1) than the sterically more hindered complex 2; therefore, subsequent mechanistic and kinetic investigations were performed with catalyst 2.
Kinetic studies with catalyst 2 showed that the reactions are first order in aminoalkene substrate and first order in catalyst (Figure 2 and 3). The activation parameters for the cyclization of 2,2-dimethylpent-4-en-1-amine were determined as ΔH# = 10.5 ± 0.4 kcal mol–1 and ΔS# = 45.7 ± 1.4 cal K–1 mol–1. Using an N-deutero substrate we were able to establish a significant primary kinetic isotope effect of 3.9 for this catalyst system. Batch-wise addition of substrate also suggests that the catalyst system exhibits product inhibition. An important mechanistic observation is the fact that the cyclization does proceed only if more than one equivalent of aminoalkene substrate (with respect to the magnesium catalyst) is present in the reaction mixture, thus indicating that two substrate molecules are bound to the catalytic metal center in order for the reaction to proceed.
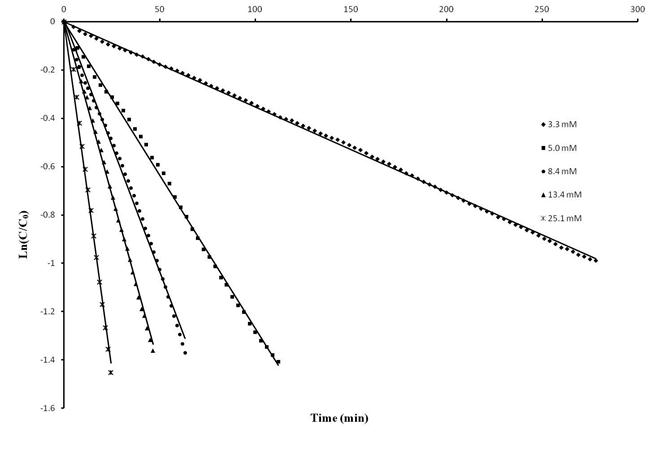
Figure 2. Time dependence of substrate concentration in the hydroamination/cyclization of (1-allylcyclohexyl)methylamine ([subst.]0 = 0.167 mol L–1) in C6D6 at 25 °C using varying concentrations of 2. The reaction is first order in substrate.
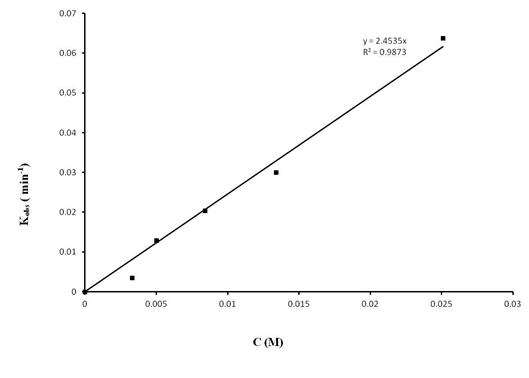
Figure 3. Dependence of catalyst concentration on observed rate for the hydroamination/cyclization of (1-allylcyclohexyl)methylamine ([subst.]0 = 0.167 mol L–1) with catalyst 2 in C6D6 at 25 °C. The reaction is first order in catalyst.
The DFT study performed by Dr. Tobisch at the University of St Andrews on catalyst system 1 in the cyclization of 2,2-dimethylpent-4-en-1-amine revealed that the two-step insertion/protonolysis mechanism prevails over a concerted mechanism. The insertion appears to be reversible with an activation barrier of 10.4 kcal mol–1 (Figure 4a). The subsequent protonolysis is rate-determining with an activation barrier of 23.6 kcal mol–1 (Figure 4b). A concerted N-C ring-closure/protonolysis pathway can be discarded due to a significant higher activation barrier of 29.7 kcal mol–1 (Figure 4c).
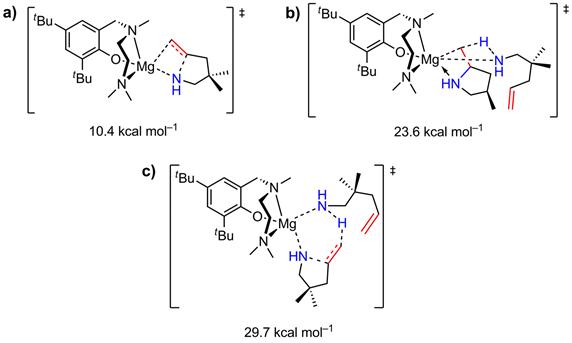
Figure 4. Computed transition state energies: a) migratory olefin insertion into the Mg-N amido s-bond; b) Mg-C protonolysis; c) non-insertive N-C ring closure with concurrent amino proton transfer onto the olefin.









