Reports: ND451429-ND4: Photochemical Reduction of Carbon Dioxide using Organic Photocatalysts
 printer friendly
printer friendlyThe objective of this project is to characterize innovative organocatalysts for the photochemical reduction of carbon dioxide (CO2). Successful realization of this goal will benefit the long-term efforts to mitigate the effects of greenhouse gas emission. However the efforts from this grant focus on identifying methods for harnessing light energy to accomplish initial one or two electron reductions of CO2. Such processes are expected to produce formic acid and/or oxalic acid. Further reduction of these products to useful fuels such as methanol or methane, coupling the CO2 reduction to the splitting of H2O, and efficiently harvesting solar energy are also also interesting and significant challenges, but these are beyond the scope of the current study.
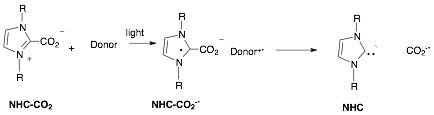
To date our efforts have focused on the use of N-heterocyclic carbenes (NHCs) as catalysts. Louie et al. have shown that a variety of these species can trap CO2, reversibly forming NHC carboxylate zwitterions (NHC-CO2). It was our hypothesis that one-electron reduction of NHC-CO2 derivatives would produce the corresponding anion radical (NHC-CO2-¥). Preliminary DFT calculations suggested that the latter was thermodynamically capable of splitting to reform the NHC along with the CO2 anion radical. The latter would not be stable, but could either dimerize to produce oxalate dianion, or abstract a H atom from the solvent (or other H atom donor), creating formate anion (HCO2).
The initial step in this overall photochemical process was envisioned as an excited state electron transfer reaction from an electron donor to the NHC-CO2 . The thermodynamic driving force (�G, in kcal/mol))for this step is determined by the reduction potential (Ered in V) of the acceptor (NHC-CO2), the oxidation potential (Eox) of the donor and the energy available from excitation (Eoo in kcal/mol) as shown in eq 1 (S is a solvation term which is generally negligible in polar solvents). As rule, excited state electron transfer steps have to be exergonic in order to occur with any efficiency. Unfortunately the reduction potentials of the NHC-CO2 species were unknown at the outset of the project.
�G=23.06Eox-Ered-Eoo-S![]()
(1)
Our first efforts were to characterize the previously synthesized N,N-dimethylimidazolium carboxylate. While we were unable to determine its Ered value electrochemically, we did discover, through fluorescence quenching experiments, that this derivative can accept electrons from excited state donors such as N,N,N',N'tetramethylbenzidine (TMB), 2-naphthoate anion (2-nap) and 2-phenanthrolate anion. On this basis, we infer that the reduction potential of the dimethyl derivative is more negative than -2.8 V (vs. SCE).
Having established the feasibility of the initial step, attention was turned to the nature of the bond-breaking step. Specifically, preparative photolysis of the dimethyl derivative was carried out using various donors, light sources, solvents and additives. The products were then examined using 1H and 13C NMR. The specific objective was, first, to determine if the electron transfer process resulted in any net chemistry, then, secondly, to determine if the following chemical processes result in the net reduction of CO2. With regard to the former, it is important realize that any bond breaking processes that follow a photoinduced electron transfer step need to compete with back electron transfer reaction, which restore the reactants with a net production of heat. With regard to the second issue, the desired bond breaking process would produce NHC, along with CO2-¥. However the latter is not stable and the eventual outcomes could include a secondary H atom transfer, creating formate ion, dimerization to create oxalate ion, or reduction of the oxidized donor resulting in the formation of CO2 and NHC. (the NHC, in turn, can get protonated to form the imidazolium ion NHC-H+).
Our current findings show that photolysis of NHC-CO2 in CH3CN/H2O solutions using either TMB or 2-Nap as the excited state donor produces formate ion along with NHC-H+. In analyzing the peak areas for the products by 1H NMR, it is seen that the formate is formed in ca. 10-35% yield relative to the NHC-H+ by product. The other possible reduction product ,oxalate, has not yet been detected by 13C NMR.
We hypothesize that the discrepancy between the formate yield and the yield of NHC-H+ is oxidation of CO2-¥ by the oxidized electron donor. In this case, including efficient H atom donors to trap CO2-¥ before it is oxidized by sensitizer ought to increase the yields. This hypothesis is currently under investigation.
An additional question raised by our current results is why the conditions examined to date require such prolonged photolysis times (>12 h using a Xe arc lamp). This may be indicative of a C-C bond breaking rate constant that is slow relative to back electron transfer. In this case, we anticipate that weakening the NHC-CO2 bond would increase the rate of bond breaking. Previous work has shown that these bond strengths can vary dramatically with the nature of the substituents on the NHC. Current efforts also include the synthesis and characterization of NHC-CO2 derivatives beyond the N.N-dimethyl. For example we have recently prepared the N,N'-dimesityl derivative and are currently characterizing its behavior through fluorescence quenching experiments.