Reports: ND149733-ND1: Catalytic Asymmetric Incorporation of CO2: Complex Molecule Synthesis from a Renewable Source of Carbon
 printer friendly
printer friendlyCarbon dioxide is the ideal C1 source for organic synthesis because of its abundance, low cost and non-toxicity.[1] CO2 may be considered a renewable resource, which makes it an attractive subject of research due to depleting fossil fuel reserves. Using CO2 as a raw material in large scale organic synthesis is currently limited to simple building blocks such as salicylic acid, urea derivatives, methanol, cyclic carbonates and polycarbonates.35 For the synthesis of fine chemicals, CO2 has not found widespread application because reactive organometallic reagents have low functional group tolerance. Recently a number of mild, transition metal catalyzed carboxylation methods have been reported[2] that provide opportunity for inventing asymmetric variants to furnish highly valuable chiral organic building blocks. Examples for asymmetric chemistry involving CO2 are still rare.[3] We reported a mild and efficient method for the carboxylation of organozinc reagents under palladium and nickel catalysis (Fig. 1).[4]
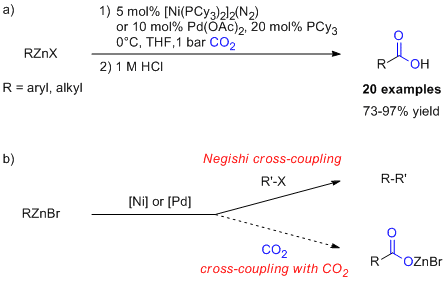
Figure 1. a) Dong's Ni-catalyzed carboxylation of organzinc bromides. b) Analogy to the Negishi cross-coupling.
We speculated that secondary organozinc reagents could be converted into valuable, enantiomerically pure products in an analogous process via (dynamic) kinetic resolution (Fig. 2). Here we present our progress with this project that included an investigation of a large number of chiral ligands (L*).
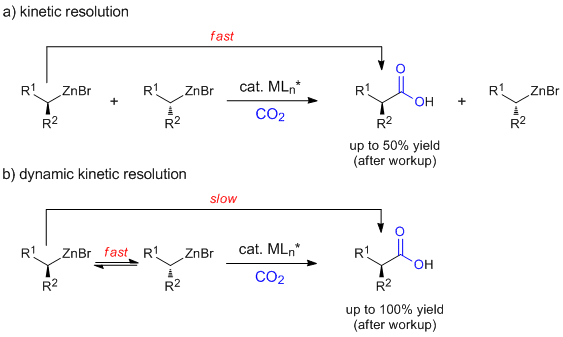
Figure 2 Proposed enantioselective carboxylation via (a) kinetic or (b) dynamic kinetic resolution.
Results and Discussion
By submitting phenylzinc bromide 7a to the previously reported reaction conditions, we were pleased to obtain an isolated yield of 62% of pure benzoic acid 8a (Fig. 3a). When secondary organozinc bromides 7b and 7c[5] were submitted to identical reaction conditions, decreased yields were observed for the corresponding acid products and for substrate 7c a mixture of isomeric products was observed. A control experiment was conducted with substrate 7b to confirm that in the absence of nickel catalyst no reaction occurs. These results indicate that secondary organozinc bromides are less reactive then primary organozinc bromides, which show slow background reactivity.
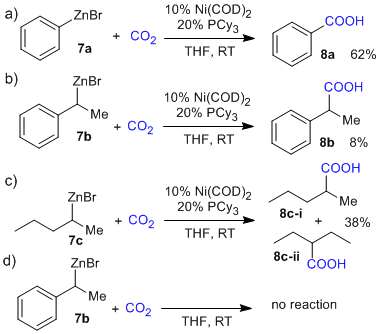
Figure 3. Reaction of primary (a) and secondary organozinc bromides (b, c) with CO2 under Ni catalysis. (d) Control experiment in the absence of catalyst.
Encouraged by a report by Biscoe[6] who used N-based ligands to suppress unwanted side reactions of secondary organozinc iodides, we investigated ligands and nickel precatalysts for the reaction of 7b with CO2 (Table 9).
Table 1. Investigation of ligands and nickel precatalysts for the carboxylation of secondary organozinc bromides
Entry | Ni source | ligand | T | 8b (%)a | eeb (%) |
1 | - | - | RT | 0 | - |
2
| Ni(acac)2 | - | RT | 7c | ND |
3
| Ni(COD)2 | PCy3 | RT | 8c | ND |
4
| Ni(acac)2 | PCy3 | RT | 26c | ND |
5
| NiCl2×DME | PCy3 | RT | 23c | ND |
6
| Ni(acac)2 | bipy | RT | 6c | ND |
7
| Ni(acac)2 | trans-1,2-diaminohexane | RT | 0 | - |
8
| Ni(acac)2 | IPr | RT | <5 | ND |
9
| Ni(acac)2 | (R)-SITCP | RT | 0 | - |
10
| Ni(acac)2 | (R)-MeDuphos | RT | <5 | ND |
11
| Ni(acac)2 | (R,R,S,S)-Duanphos | RT | 25c | <5 |
12
| Ni(acac)2 | (R,R)-QuinoxP | RT | <5 | ND |
13
| Ni(acac)2 | (R,R,S,S)-Duanphos | RT | 35c | <5 |
14
| Ni(acac)2 | 1,10-phenanthroline | RT | <5 | ND |
15
| Ni(acac)2 | (R,R)-Ph-BOX | RT | <5 | ND |
16
| Ni(acac)2 | (R,R)-Ph-PyBOX | RT | 0 | - |
17
| Ni(acac)2 | (R)-SIPHOS | RT | 0 | - |
18
| Ni(acac)2 | (R,R)-Me-BPE | RT | 12 | <5 |
19
| Ni(acac)2 | (S)-BINAPINE | RT | <5 | ND |
20
| Ni(acac)2 | (R)-ShiP | RT | <5 | ND |
21
| Ni(acac)2 | (R,R)-BenzP | RT | <5 | ND |
22
| Ni(acac)2 | (S,S,R,R)-TangPhos | RT | 24c | <5 |
23
| Ni(acac)2 | SL-J003-1 | RT | <5 | <5 |
24
| Ni(acac)2 | SL-J009-1 | RT | <5 | ND |
25
| Ni(acac)2 | SL-J002-1 | RT | 0 | - |
26
| Ni(acac)2 | 20% (R,R,S,S)-Duanphos | RT | ~20 | <5 |
27
| Ni(COD)2 | (R,R,S,S)-Duanphos | RT | 72c | <5 |
28
| NiCl2×DME | (R,R,S,S)-Duanphos | RT | ~20 | <5 |
29
| Ni(acac)2 | (R,R)-iPr-BPE | RT | 7c | ~ 10 |
30
| Ni(acac)2 | (S,S)-iPr-BPE | RT | ~10 | ~ -10 |
31
| Ni(acac)2 | (R,R)-Et-BPE | RT | <5 | ND |
32
| Ni(acac)2 | (S,S)-Ph-BPE | RT | 0 | - |
33
| Ni(COD)2 | (R,R)-iPr-BPE | RT | 0 | - |
34
| Ni(COD)2 | (R,R,S,S)-Duanphos | 50 °C | 65 | 30d |
35
| Ni(COD)2 | (S,S,R,R)-Duanphos | 50 °C | tbd | <5 |
36
| Ni(COD)2 | (R,R,S,S)-Duanphos | 50 °C | tbd | <5 |
37
| Ni(acac)2 | (R,R)-iPr-BPE | 50 °C | <5 | <5 |
38
| Ni(acac)2 | (R,R)-iPr-Duphos | RT | <5 | <5 |
39
| Pd(OAc)2 | (R,R,S,S)-Duanphos | RT | 0 | - |
a GC yield. b Determined by chiral GC. c Isolated yield. RT: room temperature. d reaction was not conducted under rigorous exclusion of oxygen. ND: not determined. tbd: to be determined.
A second control experiment showed that in the absence of added ligand, precatalyst Ni(acac)2 promoted observable reactivity (entry). All nitrogen-based ligands gave poor reactivity or inhibited the reaction completely (entries 6, 7, 14, 15 and 16). Most studied phosphorus-based ligands, as well as N-heterocyclic carbene IPr (entry 8) resulted in sluggish reactivity. An exception was electron-rich, bidentate ligand (R,R,S,S)-Duanphos, which in combination with precatalyst Ni(acac)2 gave yields above 20% (entries 11 and 13). Although (R,R,S,S)-Duanphos is a chiral ligand, we did not observe enantiomerically enriched products. Precatalyst Ni(COD)2 resulted in a yield of 72% with this ligand (entry 27). Encouraged by this result, we turned our attention towards other bidentate, electron-rich ligands and were pleased to observe that ligand (R,R)-iPr-BPE reproducibly gave low, but measurable enantiomeric excess (entries 29 and 30). This ligand and all variations thereof that we tested (entries 18, 31 to 33) exclusively gave yields around or below 10%. In a recent experiment we observed 30% enantiomeric excess when ligand (R,R,S,S)-Duanphos and Ni(COD)2 were employed at a temperature of 50 °C (entry 34). We are hoping to shed light on this promising result in the near future.
References
([1]) (a) Aresta, M.; Dibenedetto, A. Dalton Trans. 2007, 2975. (b) Sakakura, T.; Choi; J.-C.; Yasuda, H. Chem. Rev. 2007, 107, 2365.
([3]) (a) Nozaki, K.; Nakano, K.; Hiyama, T. J. Am. Chem. Soc. 1999, 121, 11008. (b) Lu, X.-B. et al. J. Am. Chem. Soc. 2004, 126, 3732. (c) Takimoto, M.; Nakamura, Y.; Kimura, K.; Mori, M. J. Am. Chem. Soc. 2004, 126, 5956.









