Reports: ND351566-ND3: Electrochemically-Controlled Alkene Binding Affinity of Surface-Immobilized Metal-Thiolate Complexes
 printer friendly
printer friendlyUsing funds from the ACS PRF award, we have been studying the reactivity of the ethylene-bound [Ru-1∙C2H4]+ complex in solution and in the gas phase. We also monitored the change in resistance of a solid-state film of the [Ru-1]+ complex as it binds with ethylene gas to form the [Ru-1∙C2H4]+ complex. The results are described below.
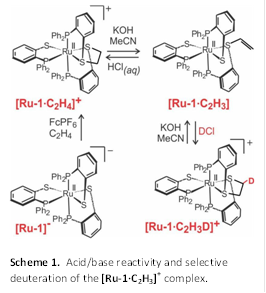
Acid/Base Reactivity. We studied the reversible deprotonation of [Ru-1∙C2H4]+ to evaluate the acid/base stability of our alkene addition complexes. Deprotonation of [Ru-1∙C2H4]+ with potassium hydroxide yields the vinyl metallosulfonium derivative [Ru-1∙C2H3] in acetonitrile via an elimination reaction in 96% yield as shown in Scheme 1. A single structural isomer with the vinyl substituent on the sulfur trans to phosphorus is obtained. The reaction does not proceed under aqueous conditions and in basic methanolic solutions consistent with the greater basicity of hydroxide in acetonitrile as compared to hydrogen bonding solvents. Acidification of [Ru-1∙C2H3] with HCl or HBr regenerates [Ru-1∙C2H4]+. The reversible deprotonation proceeds selectively at the carbon alpha to the sulfur trans to phosphorus. Deuteration of [Ru-1∙C2H3] with DCl yields the mono-deuterated dithioether complex [Ru-1∙C2H3D]+, which yields the deuterium free product [Ru-1∙C2H3] upon deprotonation. The results, corroborated by NMR investigations, indicate the selective deprotonation of a single proton in the ethylene bridge. This work has been published in the journal Inorganic Chemistry. In future studies we look to exploit this reactivity for the selective deuteration of alkenes.
Gas Phase Reactivity. We also studied the gas phase reactivity of the metal-stabilized thiyl complexes in collaboration with Professor Hao Chen of Ohio University and Dr. Larry Campbell of AB Sciex. The results support our hypothesis that the alkene addition reaction involves a metal-stabilized thiyl radical intermediate, which is a new type of distonic ion. The addition reactions of [Ru-1]+ with alkenes or methyl ketones in the gas phase were observed in agreement with the proposed mechanism. Such reactivity is also maintained by several fragment ions of [Ru-1]+, indicating that the preserved thiyl diradical core structure is responsible for the addition reaction. The thiyl radical nature of [Ru-1]+ was further verified by the ion/molecule reaction of [Ru-1]+ with dimethyl disulfide, in which the characteristic CH3S� transfer occurs, both at atmospheric pressure and also at low pressure (~mTorr). These results provide, for the first time, clear mass spectrometric evidence of the radical nature of [Ru-1]+ arising from the oxidation of non-innocent thiolate ligands of the complex [PPN][Ru-1]. The results provide support for our reaction method and an alternate method to selectively detect and identify alkenes at low concentrations by mass-spectrometry. A manuscript on this work has been accepted for publication in the distonic ion special issue of the Journal of American Society for Mass Spectrometry.
Film Resistance Changes to Ethylene Binding. A large part of this research is the fundamental study of the resistance of the [Re-1]+ complex in the with and without the bound ethylene complex. We proposed to study the resistance of this complex in both the [Re-1]+ and [Re-1]2+ oxidation states and study them as films and as part of small nanoscale metal/molecule/metal junctions. To date, we have measured the solid-state resistance of thick films of [Re-1]+ in the absence and presence of ethylene. This involved drop-cast deposition of the [Re-1]+ complex as a solid film across an array of interdigitated gold electrodes and measure of the film resistance in the presence of nitrogen first and then in the presence of different concentrations of ethylene gas in an ethylene/nitrogen mixture. We hypothesized that upon binding of ethylene to the complex, the electrons in the complex would be less delocalized, resulting in an increase in the resistance of the molecule. This is exactly what was observed in these studies. Figure 1 shows the current measured through a film of [Re-1]+ as a function of time where the film was initially in the presence of nitrogen for 100 s and then exposed to 100% ethylene for 50 s. The film was then alternately exposed to nitrogen and ethylene for 50 s intervals. As shown, the current decreased (resistance increased) by 9-10% in the presence of 100% ethylene for three different devices. Other devices often showed 4-5% resistance increases. The change in the resistance decreased as the concentration of ethylene decreased. They typically showed a resistance increase of about 0.5-1.0 % in the presence of about 3% ethylene, which was the lowest amount of ethylene so far. It was interesting that the film of the [Re-1]+ complex exhibited significant conductivity across a 5 micron electrode gap. We originally proposed to assemble the [Re-1]+ complex into a film with gold nanoparticles as cross linkers to incorporate conductivity into the film. We found that the gold nanoparticles were not necessary since the [Re-1]+ complex as a film was conductive enough to measure. We currently do not understand the conduction mechanism. We are not sure if it is ionic in nature or if there is electron hopping through the film or some other mechanism responsible. We confirmed that ethylene binding is responsible for the change in resistance by testing a film of [Re-1], which shows no resistance change in the presence of ethylene because of the very low binding constant. This suggests that ethylene binding is the source of the resistance change. We are currently trying to test the resistance change in the [Re-1]2+ complex, which has a higher binding affinity with ethylene, and we are studying the resistance change of [Re-1]+/2+ complexes incorporated in a metal/molecule/metal nanojunction. These studies should reveal potential resistance switching behavior for these complexes upon the binding of ethylene.
![Figure 1. Response of [Re-1]+ film resistance to ethylene in the solid-state.](images/abimages/Paper_12154_abstract_18957_0.png)









