Reports: DNI350460-DNI3: Low-Valent Uranium Complexes with Redox-Active Ligands for C-O Bond Activation
 printer friendly
printer friendlyIntroduction
In these studies, uranium(III) species bearing redox-active ligands were tested for their ability to cleave carbon-oxygen bonds as a model for actinide-mediated biomass processing. These studies were envisioned as part of a theoretical catalytic cycle in which C-O cleavage generates low-valent uranium oxo species, which can be further functionalized to regenerate the uranium catalyst.
Results and Discussion
i. Low-valent Uranium Compounds for the Activation of C-O Bonds
In continuation of our studies from the first funding year, two new bulky ancillary ligand frameworks have been explored on uranium, bis(trimethylsilyl)cyclopentadiene (Cp") and dimethylphenylcyclopentadiene (CpP), for the activation carbonyls in a variety of organic substrates. In analogy to Cp*2UI, Cp"2UI was generated by salt metathesis of KCp" with UI3(THF)4. Reduction of this compound in the presence of 2,2-bipyridine (2,2'-bpy) generated the uranium(III) derivative, Cp"2U(2,2'-bpy), which has a monoanionic bipyridine ligand. In the presence of benzophenone and furfuraldehyde, reduction of the carbonyl occurs by the uranium center, as evident by infrared spectroscopy. Analysis by 1H NMR spectroscopy shows analogous products to those formed with Cp*2U(2,2'-bpy) (Cp* = 1,2,3,4,5-pentamethylcyclopentadienide) that were reported during the first funding year. The resulting products, Cp"2U(OCRR'C10H8N2) (R = R = Ph; R = furanyl, R'=H), have a new bond between the carbonyl carbon and the nearest carbon of bipyridine, presumably formed via radical coupling of the reduced carbonyl with the monoanionic bipyridine (below).

Metallation of the CpP ligand was accomplished in the same manner, however in a 1:1 ratio with UI3(THF)4, forming CpPUI2(THF)x. Addition of the potentially redox-active pyridine(diimine) ligand, PDI, generated CpPUI2(PDI), in which the PDI ligand is reduced by one electron from the uranium center as evident by X-ray crystallography. Reduction with 2 eq. KC8 forms the highly reduced species, CpPU(PDI)(THF) in analogy to Cp*U(PDI)(THF) produced during the first funding year. Addition of benzophenone results in carbonyl reduction and radical coupling to form CpPU(OCPh2)2(PDI), which has a new bond between the carbons of the benzophenone ligands. Confirmation by X-ray crystallography showed a reduced pyridine(diimine) ligand in the (-1) oxidation state. The same reaction with acetone generated multiple unidentified products, indicating that perhaps the phenyl groups are responsible for radical stabilization via resonance, facilitating radical coupling. Using aldehydes does not result in any C-O bond activation, but instead makes the decomposition product, CpP2U(PDI). Reduction of CpPUI2(PDI) with 1 eq NaHBEt3 forms CpPUI(PDI), featuring a reduced PDI ligand. Further reactivity with aldehydes and ketones, including acetone, benzaldehyde, and acetophenone, is reminiscent of the Cp* and Cp" systems, as 1H NMR spectroscopic data is consistent with radical coupling of these substrates to the reduced imine arm of the PDI ligand.
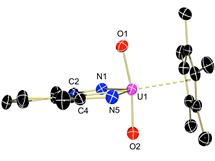
With the results of the radical coupling of reduced carbonyls in hand, we turned our attention to a previously studied system with the sterically bulky hydrotris(pyrazolylborate) (Tp*) bis(ligand) system. Using a larger ligand framework should induce loss of the redo active ligand, preventing coupling with it. Treating Tp*2U(2,2'-bpy), which has the same monoanionic bipyridine ligand as the Cp* system, with 0.2 eq glucose pentaacetate afforded Tp*2U(O2CMe) in high yields. Mass spectral data of the reaction mixture showed free bipyridine as well as methylpyran, indicating cleavage of the C-O bonds by the low-valent uranium center (below).
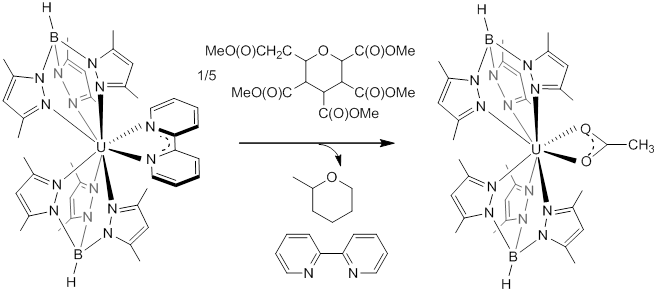
Encouraged by these results, benzophenone reduction was also successful using Tp*2U(2,2'-bpy). In this case, radical reduction of the substrate occurs, generating a rare uranium(III) benzophenone radical species, Tp*2U(OCPh2), that was spectroscopically and structurally characterized.
ii. Synthesis and Reactivity of Uranium(III) Oxos
From our previous studies, several routes for generating low-valent uranium oxo complexes were explored. In analogy to Cp*2U(2,2'-bpy) reported during the first funding year, oxidation of Cp"2U(2,2'-bpy) with pyridine-N-oxide (PNO) was achieved, generating the uranium(IV) oxo, Cp"2UO(2,2'-bpy). Due to the fact that the desired uranium(III) oxo was not obtained, a more highly reduced species, Cp*U(PDI)(THF), was oxidized instead. Treating this species with PNO in either 1:1 or 2:1 ratios resulted in formation of the dioxo species, Cp*UO2(PDI). Interestingly, a mixture of the pentavalent and hexavalent products was obtained. This difference in overall complex oxidation state is attributed to the pyridine(diimine) ligand, which we believe is found in both the neutral (0) and (-1) oxidation states, respectively, and is evident by 1H NMR spectroscopy, where both a paramagnetic and diamagnetic product result. Interestingly, while traditional uranyl species vary from red to yellow in color, both products of Cp*UO2(PDI) are blue in color, indicating that the PDI ligand is in part responsible for the unusual optical properties. In order to determine if this mixture of products is due to the oxo ligands, Cp*U(PDI)(THF) was also treated with the nitrogen analogue azobenzene (PhN=NPh). Under the same reaction conditions, the analogous bis(imido) products, Cp*U(NPh)2(PDI), were isolated, again in both the +5 and +6 oxidation states, displaying the same spectroscopic features. These bis(imido) species are rare in comparison to the dioxo derivative. Ligand oxidation states were further confirmed using X-ray crystallography for Cp*U(VI)O2(PDI) and Cp*U(V)(NPh)2(PDI). While the former species shows metrical parameters consistent with ligand reduction, the latter species show parameters as expected for a neutral ligand that compares well with uncoordinated ligand distances. These studies indicate that use of a highly reduced species does not produce a low-valent uranium oxo; instead, two oxidation events occur generating high valent products.


Conclusions
From this funding, our research has uncovered several key points:
- Uranium(III) species are capable of reducing C-O bonds. Typically radical chemistry occurs, reducing one bond at a time; double bonds are reduced to single, and single bonds are cleaved. Uranium(III) derivatives may be useful for biomass activation where single C-O bonds are common, such as glucose.
- Uranium(III) oxos have evaded isolation in these studies due to the fact that highly reduced species undergo multiple oxidation events to achieve thermodynamically favored oxidation states.
- Redox-active ligands help to mediate radical chemistry, and serve to stabilize highly reduced uranium centers for isolation of compounds that can later be used for C-O bond activation.









