Reports: DNI150350-DNI1: Regioselective Catalytic C-H Oxidation of Hydrocarbons in Aqueous Solution
 printer friendly
printer friendlyOur work in this grant period has focused on the synthesis and reactivity studies of molecules that are capable of oxidation of simple unactivated hydrocarbons. Our ultimate goal is regioselective catalytic CH oxidation is extremely rare in the literature. The catalyst not only requires good reactivity, but a recognition element must be incorporated into the catalyst to discriminate between electronically identical reactive sites. We seek to use proximity-directed reactivity to control oxidation selectivity and create catalysts that are truly biomimetic.
To create a catalyst that is capable of regioselective CH oxidation, three factors must be addressed: the catalysts must be capable of non-covalently binding the target molecule and orienting it in one direction, must incorporate a catalytic motif (usually a high-oxidation state metal species) and must allow release of the product from the binding site. Multiple metals can be coordinated to appended triazole or pyridine nitrogen ligands for catalytic activity. We focused on performing the reactions in water - by exploiting hydrophobic control of guest binding, turnover of the more hydrophilic oxidation products is possible. This, of course, requires synthesis of catalysts that are both tolerant to, and soluble in aqueous solution.
The cavitand synthesis is shown in Figure 1. Copper catalyzed azide-alkyne cycloaddition (CuAAC) reactions allow extension of the host walls. Simple reaction of cavitands 1a/b with sodium azide provides tetrazide cavitands 2a/b in good yield. Both the acetate- and alcohol-footed cavitands were synthesized to allow solubility in a range of solvents. Five different metal-coordinating groups have been introduced (and their properties analyzed) to date; for brevity we will focus on cavitands 6 and 7 here.
Figure 1. Receptor scaffolds. Synthesis of metal-coordinated water-soluble cavitands via CuAAC chemistry.
The triazole-based cavitands are capable of self-folding via metal coordination in two different conformations, with the metal positioned directly over the cavity through triazole coordination (leaving empty coordination sites positioned vertically, as in 6�Cu), or in our desired conformation where the two metals are positioned at the sides (as in 7�Fe2, Figure 1). The geometry is dependent on a variety of factors, including the functional groups appended to the triazoles and the nature of the metal species. For example, Cu(I) shows preference for coordination at the triazoles, while Fe(II) salts have a much lower affinity for this coordination mode. The metal-coordinated cavitands are water soluble, and this solubility depends on the rim and foot substitution. The greatest water solubility (>20 mg/mL) is possible if the alcohol feet are converted to sulfates, but manageable water solubility (~4 mg/mL) is observed for the alcohol-footed cavitands 6a�Fe and 7a�Fe2. The coordination of charged metal species confers water-solubility on system without covalent introduction of solubilizing groups.
Substrate | Cat. | Product Yield(s), % | |
|
|
| |
7b•Fe2 | 54 | 4 | |
6b•Fe | 67 | 12 | |
|
|
| |
7b•Fe2 | 32 | 26 | |
6b•Fe | 35 | 22 | |
|
|
| |
7b•Fe2 | 48 | 20 | |
6b•Fe | 52 | 17 | |
(n-Bu)2O |
|
| |
7b•Fe2 | 51 | 23 | |
6b•Fe | 56 | 21 | |
|
| ||
7b•Fe2 | 88 | ||
6b•Fe | 87 | ||
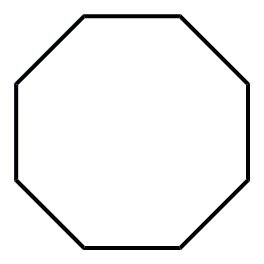
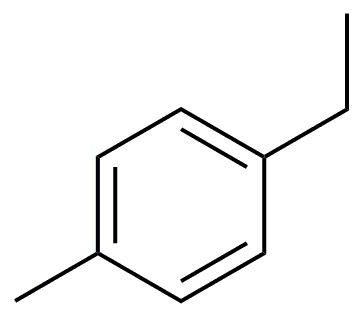
Figure 2. Catalytic hydrocarbon C-H oxidation by metallated cavitands.
Each cavitand displays a metal species with two free coordination sites that is a potential oxidation catalyst. The copper-coordinated cavitands are effective oxidation catalysts, but our most exciting results have been with the iron (II) coordinated hosts. The empty (or weakly coordinated by solvent and/or counterion) coordination sites in 6b•Fe and 7b•Fe2 allow for in situ formation of the required Fe(V) oxo species for hydrocarbon oxidation. Optimized oxidation conditions (10% catalyst, 10 eq. tBuOOH, EtCN:H2O, ~200mM), allow reaction of completely unactivated hydrocarbons (see Figure 2 for partial results). Cyclic and polycyclic hydrocarbons were good substrates for the reaction, and there was little difference in the effectiveness of cavitand catalysts 8b•Fe and 9b•Fe2. Unfortunately, linear alkanes such as n-octane were unreactive, as were branched acyclic alkanes such as 2,2,4-trimethylpentane. Selectivity was observed in certain cases: the product of cis-decalin oxidation favored the more sterically accessible C-H bonds, giving cis-9-decalol and cis-2-decalone in a 2.5:1 ratio. Trans-decalin, on the other hand, was oxidized to trans-2-decalone and trans-1-decalone in a 2:1 ratio with an overall yield of 53%. No tertiary C-H oxidation was observed. p-Xylene was unreactive, however the oxidation of 4-ethyltoluene gave 4-methylacetophenone in 88% overall yield at ambient temperature. In each of these cases, performing the oxidation with uncoordinated FeSO4 gave minimal conversion. The cavitands do not undergo observable self-oxidation or decomposition during the reaction: if the oxidation is quenched by addition of ether, the catalyst can be recovered by filtration, and still displays coordinated Fe.
In summary, we have synthesized cavitands that are water-soluble, able to self-fold upon coordination with suitable metals and perform catalytic C-H oxidation reactions of unfunctionalized alkanes with little to no degradation in aqueous solvents. The catalysts are tolerant to the oxidation conditions and are capable of challenging oxidation reactions under mild conditions. The challenge is to apply these successes to a host that displays a larger cavity for guest recognition and does not suffer from cavity blockage by the bound metal ions. In addition, greater catalytic activity is desirable to maximize the scope of the process and access more challenging targets such as linear alkanes. This work has been communicated in two publications.
Also in this grant period, we have published three further journal articles on work partially supported by this grant. These publications involve study of the attachment of deep cavitands to solid surfaces and supported lipid bilayers. The cavitand catalysts described above are excellent substrates for heterogeneous catalysis if suitable surface attachment procedures are found, and we have studied two possible methods for this process, either by incorporation in membranes or directly attached to the surface via thiol-gold contacts. These cavitand-surface conjugates have been used for a variety of molecular recognition processes including biosensing and protein immobilization. We are currently investigating the possibility of surface attachment of 6 to create a fully reusable hydrocarbon oxidation catalyst.









