Reports: ND749719-ND7: Novel Approaches to Photovoltaic Devices and Materials
 printer friendly
printer friendlyFrom medieval times, azure-blue colored natural products based on the azulene (C10H8) skeleton have attracted particular attention, initially for their physical properties, but more recently for their novel electronic structures. Significantly, the parent molecule, azulene is a 10 p-electron isomer of naphthalene, yet it exhibits a dipole moment of 1.08 D and a deep blue color. While unusual for small unsaturated aromatic hydrocarbons, these properties result from the fusion of an electron-rich five-membered ring and electron-poor seven-membered ring. This remarkable electronic structure of azulene allows for cation stabilization through aromatization of the seven-membered ring, which may be exploited in advanced materials for electronic, optoelectronic and electrochromic devices.
The traditional synthetic approaches to these electronically unique building blocks are characterized by long and elaborate synthetic procedures that are low yielding and in many cases do not afford the desired substitution patterns. Herein we report a versatile and modular strategy for the synthesis of azulene derivatives having a single isomeric arrangement of reactive functional groups in the seven-membered ring and the use of these functional azulenes as building blocks in order to construct stimuli-responsive oligomers.
The requirement for the regiospecific introduction of reactive functional groups to the seven-membered ring of azulene necessitated the development of a new synthetic strategy for these building blocks. To address this challenge, the 6p+4p cycloaddition of thiophene-S,S-dioxides and fulvenes was examined (Scheme 1). This reaction allows the synthesis of the azulene nucleus in a single step with the substitution pattern of the seven-membered ring being derived from the original thiophene ring. Another advantage of this strategy is the availability of a wide variety of substituted thiophene-S,S-dioxide derivatives through the use of the acetonitrile complex of hypofluorous acid (HOF.CH3CN), which is a strong, yet selective oxidizing agent. For this work, a freshly prepared solution of HOF.CH3CN was used to oxygenate 2,5-dibromothiophene 1a, 2,5-dibromo-3-dodecylthiophene 1b, and 2,5-dibromo-3-methylthiophene 1c, leading to the corresponding 2,5-dibromothiophene-S,S-dioxide 2a and the previously unknown alkyl-substituted thiophene-S,S-dioxides, 2b and 2c, in high yield (Scheme 1).
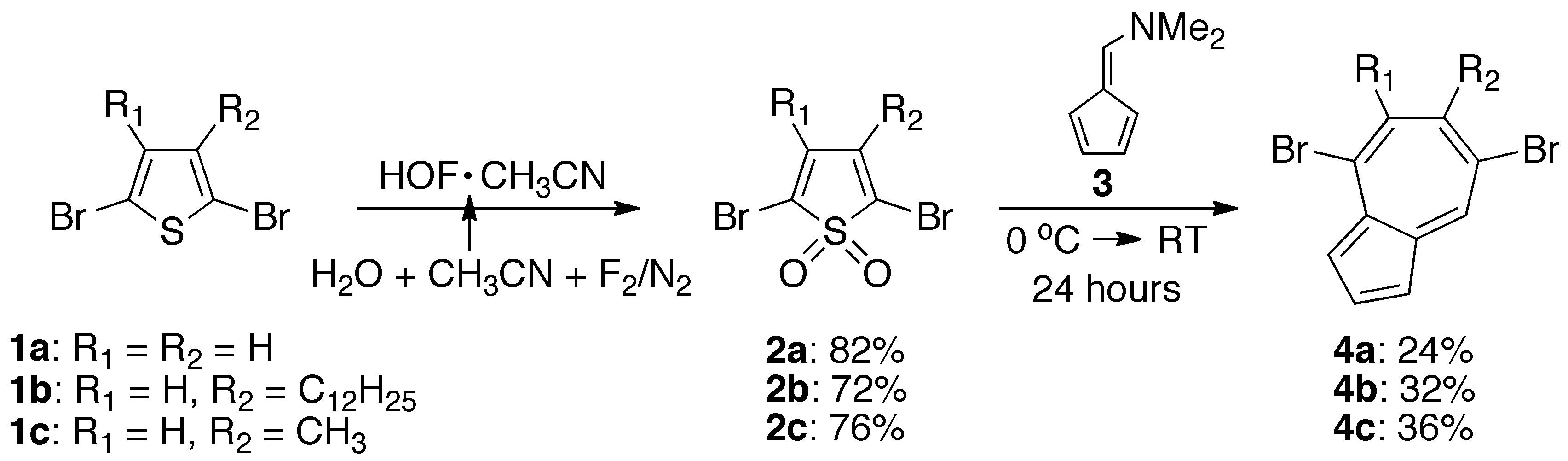 |
Scheme 1. Synthesis of substituted azulenes 4a, 4b and 4c.
Examination of the cycloaddition reaction between thiophene-S,S-dioxide 2a and dimethylaminofulvene 3 revealed that both the reaction conditions and the sequence in which the compounds are introduced are important for increasing the yield of 4,7-dibromoazulene 4a (Supporting information). Similarly, the reaction between sulfone 2b and dimethylaminofulvene 3 gave previously unknown azulene derivative 4b, substituted with two bromines and a dodecyl group at the seven-membered ring (Scheme 1). The presence of the dodecyl chain is important to improve the solubility of oligomers and polymers containing azulene 4b.
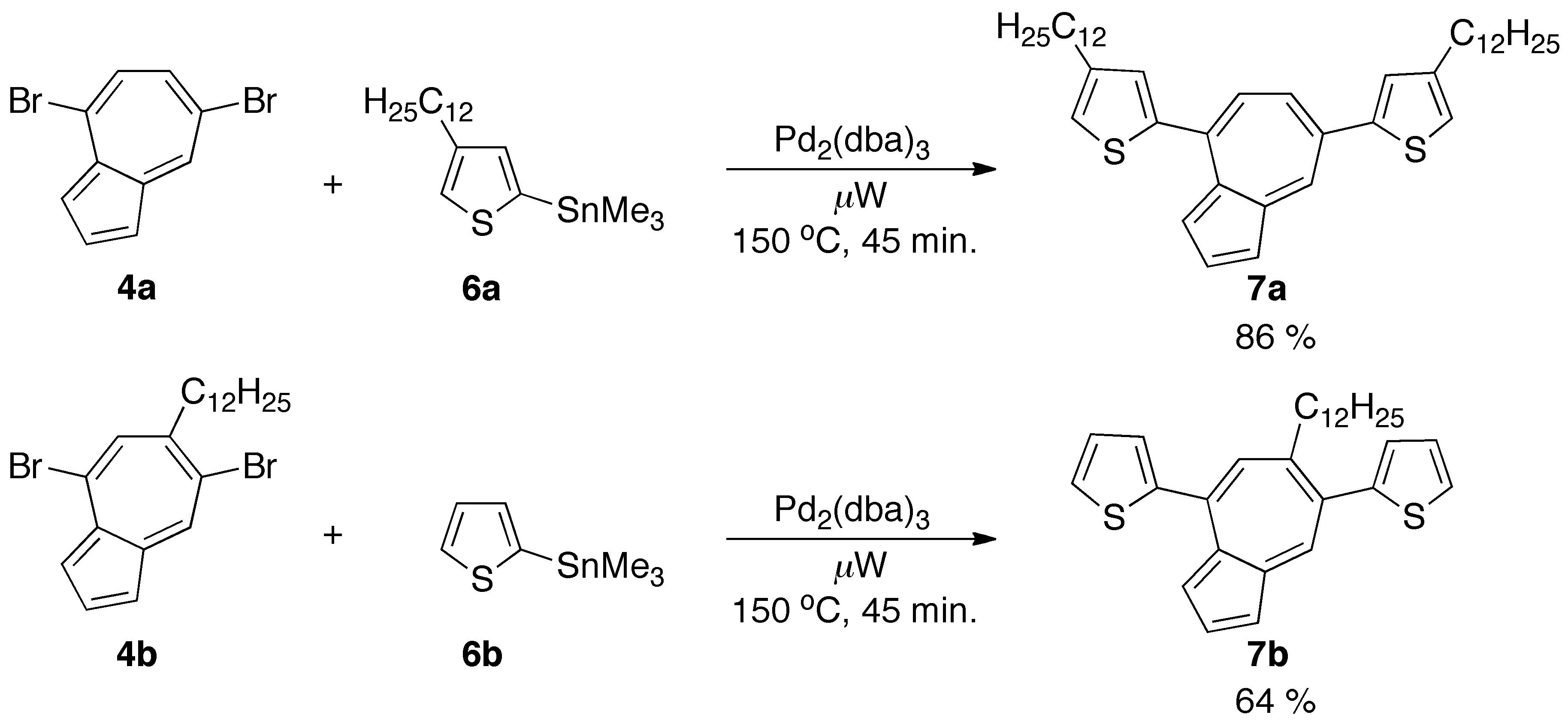
Treatment of compounds 4a and 4b with trifluoroacetic acid (TFA) resulted in the formation of a complex mixture of products, most likely due to instability of the generated species (Supporting Information). In order to stabilize the azulenium cation, we designed conjugated oligomers based on the substitution of the seven-membered ring of the azulenes with thiophenes. Accordingly, 4,7-dibromoazulenes 4a and 4b were reacted with 2-(trimethylstannyl)thiophenes 6a and 6b respectively via microwave-assisted Stille cross-coupling reaction. Novel azulene-thiophene oligomers 7a and 7b were isolated as blue solids in good yield (Scheme 2).
Scheme 2. Synthesis of azulene-thiophene oligomers 7a and 7b.
The formation of azulenium cations from the conjugated azulene-thiophene oligomers, 7a and 7b, was then examined. Upon addition of TFA to a dichloromethane solution of 7a, protonation of the five-membered ring of azulene takes place generating a mixture of azulenium cations, 8 (Figure 1). This is accompanied by an instant color change of the solution from deep blue to orange-red. 1H-NMR shows that the proton signals in the aromatic area shift, and a new peak appears at 4.2 ppm corresponding to the CH2 protons of the protonated five-membered ring. Significantly, the formation of the azulenium cations proved to be a reversible process with oligomer 7a being regenerated by addition of base, such as triethylamine or by evaporation of the solvent and the acid. It is important to note that this process of protonation and deprotonation of the oligomer could be repeated multiple times (> 10) with no observed degradation of the oligomer, demonstrating the stabilization of the azulene nucleus through conjugation with the thiophene rings. Furthermore, the protonation is not limited to TFA and can be achieved utilizing different acids, such as perchloric, hydrochloric, and sulfuric acids with similar results being obtained for the oligomer 7b.
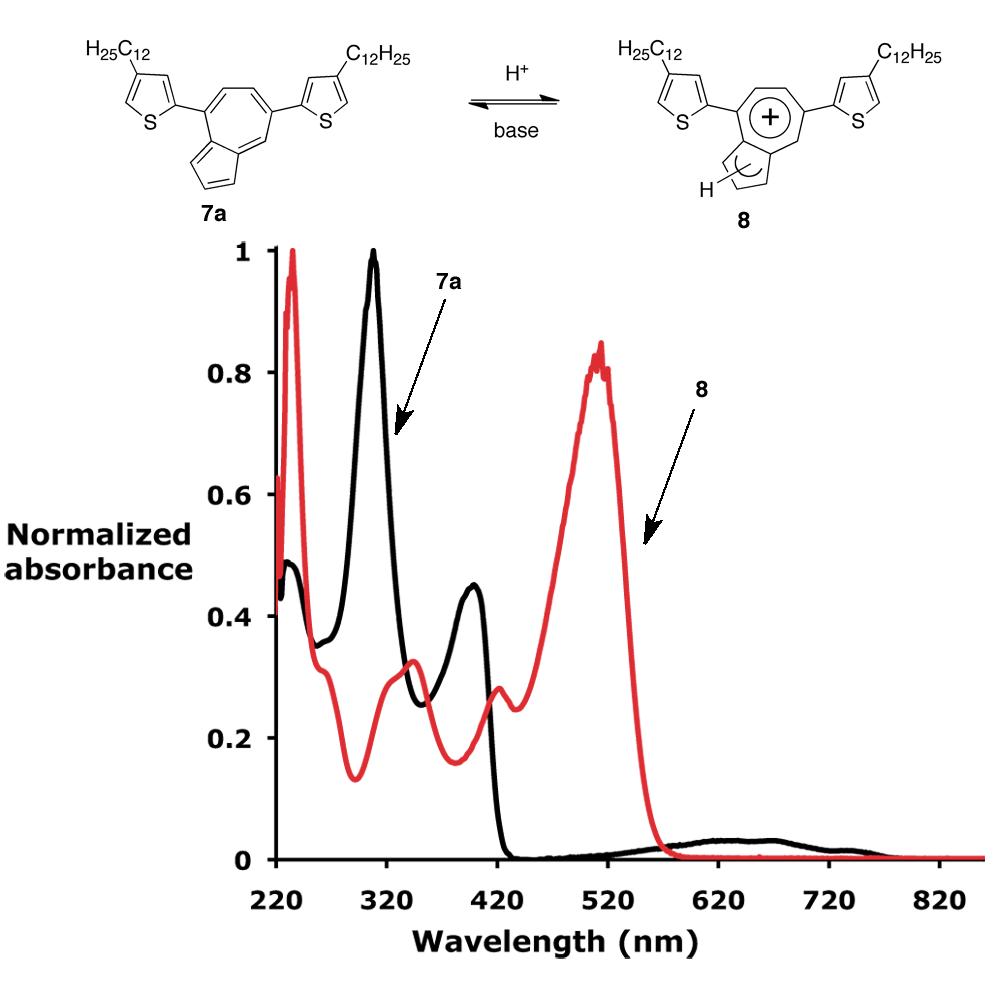
Figure 1. Top: Formation of azulenium cations, 8, from thiophene-azulene oligomer 7a. Bottom: UV-vis measurements in dichloromethane for the oligomer 7a in its neutral form and after protonation, 8.
Comparison of the neutral azulene derivative, 7a, with the derived azulenium cations, 8, showed a significantly smaller energy gap for 8 as evident from the UV-vis spectra (Figure 1). The spectrum of 7a exhibits two major maxima at 308 nm and 402 nm corresponding to π-π* transitions of the azulene and thiophene rings, respectively with a weak broad absorbance between 615 and 640 nm due to the S0-S1 transition of the azulene unit. Upon protonation, a new peak at 514 nm, which is attributed to the azulenium cation, is observed and the efficient conjugation of the azulenium cation with the adjacent thiophene rings is evidenced by this absorption maximum being red-shifted significantly (Dlmax = 162 nm), with respect to the values reported in the literature for the unsubstituted azulenium cation (352 nm).
In conclusion, access to novel azulene derivatives could be obtained through the regiospecific cycloaddition of thiophene-S,S-dioxides with fulvenes and subsequent cross coupling with trimethylstannyl thiophenes. In a reversible process, the optical band-gap of these azulene-thiophene oligomers could be easily modulated by simple protonation with fluorescence being "switched on" upon generation of the corresponding azulenium cations. The presence of the thiophenes is essential for stabilizing the azulenium cations and the modularity of our approach allows other substituents to be explored in order to tune the properties of these novel material systems.









