Reports: UR449182-UR4: New Directions in Understanding Conformational Preferences of 1,2-Disubstituted Ethanes
 printer friendly
printer friendly1. Software is being developed to convert measured dipolar and secular proton and 13C -carbon couplings observed in either liquid-crystal nematic phases or lyotropic solutions to obtain the molecular coordinates and dihedral angles of organic compounds of the general formula X-CH2-CH2-Y, where the X and Y substituents can be the same or different. The coordinates so obtained can be straightforwardly converted to various types of molecular images. The software written in Python 7 is quite complex and a very substantial effort is being made to develop it for the purpose, which will be as understandable and user friendly as possible. The overall process is analogous to the equivalent of a crystal structure, but obtained from NMR spectra.
2. Much research has been done in the past on conformational analysis on substituted cyclohexanes and many regularities have been found as respects to conformational preferences chemical reactivities and other properties of interest. We have been interested in interactions between substituents on cyclohexane rings and especially intramolecular hydrogen bond formation. In this connection, an interesting possibility is to see if the monanions of cis-1,3- and trans-1,2-cyclohexanes behave like the monoanion of succinic acid in being barely, if any intramolecular hydrogen bonded at all in water, and yet in DMSO it is very strongly hydrogen bonded up to at least 130° C.
3. One of the most mystifying effects, we have observed in many years of study of the conformational analysis (first published in 1990) s quite simple but quite inconsistent with the things that seem now more easily rationalized. It involves the conformational preferences of a very simple system of the X-CH2-CH2-Y type where X and Y are the simple carboxylate groups so that the interest is in the dianion of succinic acid, -O2C- CH2-CH2-CO2- Conformational analysis suggests that this substance will exist in the same two conformations trans and gauche as expected for X-CH2-CH2-Y systems and its NMR spectrum, looks normal, but if physical organic chemists what the conformational preferences they would expect, they will say if you look at the Newman projections, as shown below, you will see that in the trans conformer, X and Y will be as far apart as they can be. On the other hand, the two gauche conformers have the X and Y groups much closer together. If nothing else is different, and X and Y are both negatively charged carboxylate groups, then the charges are expected to move to get farther apart and unless there is some unusual reason for the gauche to be more stable, the trans form is expected to be the more stable.
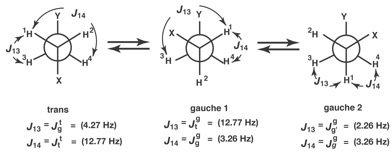
So then we take the NMR spectrum, let us say in water as the solvent, and extract the Jij coupling constants numbered as shown above and with typical values and Jijt belonging to the trans and Jijg to the gauche. Analysis of the couplings by standard software, says that the gauche preference is close to 50%, which comes as a surprise if you are really expecting it will be mostly trans.
In general, we expect that the observed preferences will be expected to change at least somewhat when we change the solvent. A common often large change is going from water to DMSO. Thus in the protic solvent water, the monoanion of succinic acid has a very small tendency to show a preference for the gauche conformer, it exists essentially at the statistical preference of two gauche to one rrans. Now, if we change to the aprotic solvent, DMSO, it changes to about 98% gauche and it is very stable at that figure even up to 130°C in DMSO.
So, now let us try going for protic water (~50% gauche for water) for the dianion, using aprotic DMSO and it turns that about the same preference (~45% gauche) as with water. Clearly, the same rules are not in play, as with the monoanion, where the change from water to DMSO is a complete turn-around. Usually simple X-CH2-CH2-Y compounds where X and Y are appropriate groups show tendencies that are similar to the mono- and dianions outlined above, but while we do understand the dramatic change of the monoanion for water to DMSO. It is harder to understand the lack of the same change for the dianion going from water to DMSO. After much thinking on this problem we decided to look and see what happened with other aprotic solvents and looked at 32 different ones from nitrobenzene to alkyl ethers and the actual substrate was the dianion with two equivalents of tetrabutylammonium results as the required cation. Again the results were very surprising, the preferences for gauche ranged from ~5% for THF and ~98% for propylene carbonate, the spread for a given solvent being about +/- 2%, or less with groups of related compounds like alcohols, esters. tert-amines, etc. with intermediate gauche values. This problem needs much more investigation and we expect to keep trying to solve it!









