Reports: DNI650645-DNI6: Intermolecular Non-Covalent Interactions in pi-Conjugated Heterocyclic Oligomers
 printer friendly
printer friendlyThe overarching goal of our ACS-PRF sponsored research program is to study the effects of intermolecular non-covalent interactions on the physical and electronic properties of π-conjugated heterocyclic oligomers. The goal is to understand how these non-covalent interactions can be exploited to tune to solid-state structures and properties of organic electronic materials. At the heart of this work is uncovering how substituents and heteroatoms alter the non-covalent π-stacking and XH/π interactions that are operative in the solid state. During this grant period, in addition to other work (vide infra) we have (1) carried out detailed studies of inter-chain interactions on the physical properties of oligothiophenes; (2) uncovered novel means of controlling the local orientations of stacked discotic systems; and (3) carried out a comprehensive study of the physical nature of substituent effects in XH/π interactions (CH/π, OH/π, etc).
Torsional Potentials of Oligothiophenes
We examined the torsional potentials of oligothiophenes and the impact of neighboring chains on these torsional potentials. Because torsional motions of conjugated oligomers can significantly affect charge transport properties, understanding the factors that impact the energy required for torsional motion will be important in the design of next-generation organic electronic materials. The aim of our work is to shed light on the rigidity of oligothiophenes in the solid state, and help bridge the gap between previous studies of isolated oligothiophene chains and their physical properties in more realistic environments.
Initially, we utilized density functional theory (DFT) to study the torsional potentials of a series of oligothiophenes (nT, n = 2-10), to examine how the torsional potentials change with increasing chain length. For sexithiophene (6T), we also predicted torsional potentials for stacked chains in several prototypical configurations to understand how a single neighboring chain impacts predicted torsional potentials. This was followed by a combined quantum mechanical/molecular mechanical (QM/MM) study of a single 6T chain in the environment of the 6T crystal structure. It was shown that neighboring chains have a significant impact on computed torsional potentials, and we suggest that studies of torsional potentials of conjugated heterocyclic oligomers should account for the effect of interchain interactions to provide an accurate portrayal of these systems.
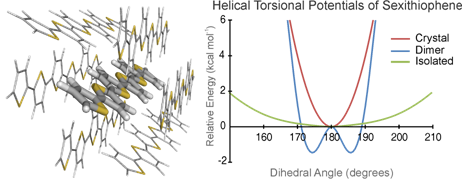
In subsequent work, which will be submitted shortly, we have performed high-accuracy ab initio computations of torsional potentials of bithiophene. Highly accurate potentials were predicted based on the Focal Point Approach of Allen and co-workers, in which one executes dual extrapolations to obtain energies approaching the non-relativistic Born-Oppenheimer limit. These reference potentials were then used to benchmark a large number of DFT functionals and basis sets to identify functional/basis set combinations that yield accurate torsional and interaction potentials at a reasonable computational cost. Surprisingly, these studies reveal no currently available DFT functionals accurately describe the torsional potentials of bifuran, bithiophene, and biselenophene. Moreover, many commonly employed functionals and basis sets yield qualitatively incorrect potentials for these systems. This has important implications for the myriad computational studies performed previously in which molecular mechanics (MM) potentials were parameterized to reproduce these DFT potentials. These accurate benchmark potentials can now be used to directly parameterize highly accurate MM potentials for molecular dynamics simulations of real conjugated organic materials.
Controlling Local Orientations in Discotic Systems
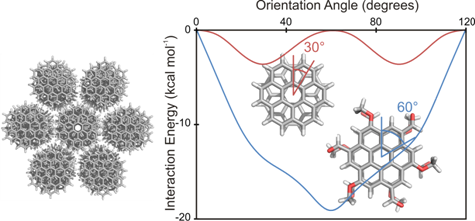
Discotic liquid crystals, which typically comprise substituted polycyclic aromatic cores that self-assemble into columnular superstructures, constitute a new generation of organic semiconductors. The fluidity and potentially high charge-carrier mobilities of these systems coupled with anisotropic mechanical and optical properties present some advantages over crystalline organic electronic materials. Two key determinants of charge-carrier mobilities in the discotic phase are the intracolumnular order and the stacking distance between the aromatic cores. That is, charge transfer rates are maximized in well-ordered, closely stacked systems. Designing systems that self-assemble into such ordered columns has proved challenging. We showed that simple substituents can have a dramatic impact on the orientation of stacked PAHs through the local, direct interactions of substituents in π-stacking interactions. For example, for alternating OMe and CHO substituents, the minimum energy stacked geometry for coronene and HBC, which adopt staggered configurations in the unsubstituted cases, feature an eclipsed structure featuring through-space local dipole-dipole interactions between the OMe and CHO groups. This orientation coincides with the maximum of charge transfer rates for such stacked systems. Such effects can provide a complementary means of controlling the orientation of π-stacked PAHs for applications in discotic liquid crystals.
Understanding Substituent Effects in XH/π Interactions
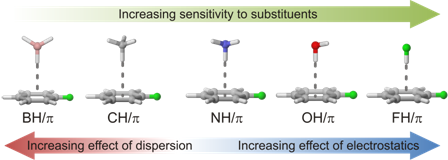
In the solid state, conjugated oligomers exhibit diverse non-covalent interactions, including CH/π interactions. Although there have been numerous computational studies of the sundry factors that influence the strength of CH/π and other XH/π interactions, to date there has been no comprehensive study of substituent effects across the full range of XH/π interactions (BH/π, CH/π, NH/π, OH/π, and FH/π). We recently carried out such a study, considering a large number of substituents and treating all five XH/π interactions at the same level of theory. This allowed direct comparison of the impact of substituents across these different interaction. Overall, substituent effects in XH/π interactions are in general accord with previous work, and follow expected trends based on simple physical models. That is, for highly polar XH/π groups (NH, OH, and FH), the impact of substituents is driven by electrostatic effects. For the non-polar groups, substituents effects are dominated by differences in dispersion interactions. Examining the full range of XH/π interactions has provided a comprehensive and unified view of the physical origin of substituent effects in these interactions.
In other work during this period, we characterized the non-covalent interactions operative in the protonated benzene dimer, quantified the interactions between natural DNA bases and a DNA isostere, and developed a hierarchy of homodesmotic reactions for accurate determination of hydrocarbon radical thermochemistry. We also published reviews on substituent effects in non-covalent interactions with aromatic rings and on a hierarchy of homodesmotic reactions for computational thermochemistry. Finally, working with colleagues in the Department of Mathematics, we wrote a paper on the physics of greenhouse gas molecules for mathematicians, in order to spur research concerning greenhouse gases and the problem of global climate change.









