Reports: DNI151247-DNI1: The Development of Enantioselective Friedel-Crafts Reactions to Prepare Unnatural Amino Acid Derivatives
 printer friendly
printer friendlyTryptophan and unnatural tryptophan derivatives are important building blocks for the total synthesis of natural products, as well as the development of new drugs, biological probes, and chiral small molecule catalysts. As part of our research program aimed at establishing new methods for the enantioselective synthesis of alkaloids, we became interested in developing convergent syntheses of tryptophans and cyclo-tryptophans (also known as pyrroloindolines) from simple indole starting materials. Our research funded by this Doctoral New Investigator award from the ACS PRF has resulted in the first direct, enantioselective synthesis of tryptophan derivatives by a tandem Friedel–Crafts conjugate addition/asymmetric protonation reaction. These reactions require no pre-activation of the indole substrates, and provide convergent access to a range of substituted tryptophan derivatives in enantioenriched form.
In 2010, we reported a new reaction for the preparation of enantioenriched pyrroloindolines (3) in which (R)-BINOL¥SnCl4 catalyzes a formal (3 + 2) cycloaddition reaction between 1,3-disubstituted indoles (1) and benzyl 2-trifluoroacetamidoacrylate (2a) (Figure 1, a).[1] Good yields, moderate exo:endo diastereoselectivities, and high enantioselectivities were obtained for a variety of indole substrates. Unexpectedly, our studies revealed that the initially formed exo- and endo-diastereomers of 3 were generated in opposite enantiomeric series. These findings led us to propose that pyrroloindoline formation proceeds by a stepwise mechanism, in which an initial conjugate addition of indole 1 to 2a is followed by a highly selective catalyst-controlled protonation to give 5. Subsequent cyclization of the amide onto the iminium ion provides the pyrroloindoline product (3).
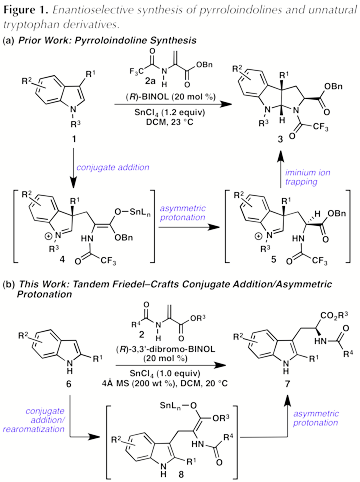
We hypothesized that a (R)-BINOL¥SnCl4 complex serves as a chiral Lewis acid-assisted Brznsted acid (LBA)[2],[3] to effect an asymmetric protonation of the enolate intermediate. Although Yamamoto and coworkers initially developed (R)-BINOL¥SnCl4 as an LBA to effect enantioselective protonation of silyl enolates, these complexes had never previously been used in tandem conjugate addition/asymmetric protonation reactions. We hypothesized that the combination of (R)-BINOL and SnCl4 could catalyze the reaction of 2-amido acrylates with C2-substituted indoles to provide unnatural tryptophan derivatives. Key to this transformation is a catalyst-controlled protonation to set the absolute configuration of the key a-amidoester stereogenic center (Figure 1, b).
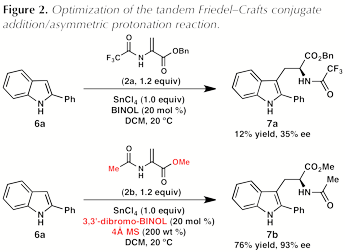
To test this hypothesis, 2-phenylindole (6a) and benzyl 2-trifluoroacetamidoacrylate (2a) were subjected to the reaction conditions previously optimized for the enantioselective formal (3 + 2) cycloaddition reaction. Somewhat surprisingly, the desired reaction was sluggish under these conditions: after 2 hours, trifluoroacetamido ester 7a was formed in low yield and poor enantiomeric excess (Figure 2). In an effort to improve the reactivity, a screen of additional 2-amidoacrylates was conducted. Gratifyingly, the use of commercially available methyl 2-acetamidoacrylate (2b) gave substantially improved results, providing acetamido ester 7b in 73% yield and 78% ee (not shown). A further survey of additives and BINOL derivatives revealed that use of 3,3'-dibromo-BINOL as the catalyst and 4 molecular sieves as an additive provided 7b in 76% yield and 93% ee.
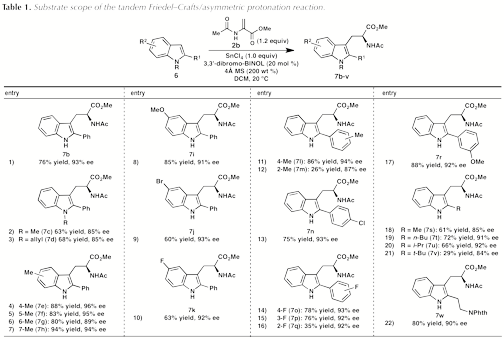
Having identified conditions to prepare acetamido ester 7b in high yield and enantiomeric excess, a survey of indole substrates was conducted to evaluate the scope of the reaction. Substitution of the 2-phenylindole backbone at the 4, 5, 6, and 7-positions is well tolerated (entries 4–7). Whereas substrates bearing either electron-donating or electron-withdrawing substituents furnish products with high enantioselectivity, the more electron-poor indoles are less reactive and provide lower yields of the acetamido ester products even with 1.6 equiv of SnCl4 (entries 9 and 10). A range of substituents are tolerated at the 2-position of the indole, including both aryl and alkyl groups. 2-Arylindoles bearing substituents in either the m- or p-position of the arene are accommodated; on the other hand, o-substituted arenes are substantially less reactive (entries 12 and 16). For indoles containing 2-alkyl substituents, the ee improves in switching from a methyl group to the slightly larger n-butyl and i-propyl substituents (entries 18-20); however, both the yield and selectivity are diminished in the case of bulky t-butyl substitution (7w, entry 22).
Importantly, this award has provided these students with the opportunity to conduct exciting research in the area of asymmetric catalysis. Several members of my group have worked on various aspects of this project, and it has proven to be a fertile area for both mechanistic studies and synthetic applications. The support provided by this DNI award has positively impacted my career by providing me with the opportunity to pursue exciting, fundamental research in the area of asymmetric catalysis. I expect that the findings described here will continue to drive an exciting area of research in my laboratory for many years to come.
References:
([2]) (a) Ishihara, K.; Nakashima, D.; Hiraiwa, Y.; Yamamoto, H. J. Am. Chem. Soc. 2003, 125, 24. (b) Nakamura, S.; Kaneeda, M; Ishihara, K.; Yamamoto, H. J. Am. Chem. Soc. 2000, 122, 8120. (c) Ishihara, K.; Nakamura, S.; Kaneeda, M.; Yamamoto, H. J. Am. Chem. Soc. 1996, 118, 12854. (d) Ishihara, K.; Kaneeda, M.; Yamamoto, H. J. Am. Chem. Soc. 1994, 116, 11179.









