Reports: ND149668-ND1: A Systematic Evaluation of Chiral Phase Transfer Catalysts
 printer friendly
printer friendlyProgress Report
I. Statement of Objectives. The primary objective of this application was to elucidate the factors that govern the structure-reactivity-selectivity relationships in asymmetric phase transfer catalysis (ATPC) with chiral quaternary ammonium ions. This program will involve an iterative process that involves the synthesis, evaluation, physicochemical analysis, and design of the chiral quaternary ammonium ions. These activities will be repeated for the optimization of the catalyst structure.
The first phase of the program involved the design and development of two different chiral scaffolds for the quaternary ammonium salts for application in asymmetric phase transfer catalysis. The [4+2] cycloaddition and tandem [4+2]/[3+2] cycloaddition of nitroalkenes are highly convergent processes that allow the assembly of rigid, polycyclic amine scaffolds that can be embellished with a variety of different functional substituents in a convergent region of space. Two different chiral scaffolds will be investigated that present substituent in a different arrangement and introduce them in a different sequence. Parallel synthesis methods will be employed to generate large libraries of ammonium ions for testing.
The chiral ammonium salts will be evaluated for their potential to catalyze a number of synthetically important transformations and a Quantitative Structure-Selectivity Profile will be developed to explain the roles of the different substituents.
II. Cyclopentapyrrolizidinium Library Synthesis and Data Collection. In May 2011, back-to-back Feature Articles were published that detailed over 5 years of work (ca. 100 journal pages and > 600 SI pages) on the synthesis, evaluation, and QSAR/QSSR modeling of cyclopentapyrrolizidinium catalysts. Over 160 ammonium salts were synthesized that contained varying numbers of substituents at positions R1-R4 as well as different configuration at C(1). The substituents allowed various effects to be evaluated such as steric and electronic contributions, p-surface, lipophilicity, and polar surface area on the catalyst structure. The rate data for the O'Donnell benzylation (50% aq. KOH/toluene) covers four orders of magnitude of activity (t1/2 from 5 min to over 15 h) and was deemed suitable for model development. In addition we determined the stir-rate dependence over a range of stir rates (1038 – 2500 rpm). Importantly, there is a sufficient range of stirring speeds, along which the activities of all of the catalysts surveyed are dependent on mixing rate (i.e. operate in the transport-limiting regime).
The enantioselectivity for the chiral catalysts was largely dependent on the substituents R2 and R4. The enantioselectivity is greatest for catalysts that bear (1) strongly electron withdrawing groups on the nitrogen, (2) a p-surface at the R2 substituent, and (3) an alkyl group at R1.
Scheme 1

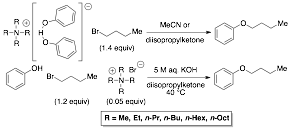
III. Reaction Rate Model Development. From the rate data collected from the library of 160 ammonium salts, we were able to develop a two-parameter model that explained a range of over 2000 fold rate difference. The two determining factors are (1) cross sectional area (XSA) and (2) solvent accessible ammonium positive charge. The balance of these two factors was able to reproduce the simple parameter "q" introduced by Halpern to correlate the rate of alkylation catalyzed by tetraalkylammonium ions. Most importantly, however, our parameters are not limited to simple quaternary ammonium salts, but can be calculated for any ammonium ion. Thus, we were able to fit our data to a parabola that relates the rate of the catalyzed alkylation with the cross sectional area of the catalyst, Chart 1. The catalyst set we prepared, however, did not contain catalysts with very large cross-sectional areas, so the descending part of the parabola has not been defined. Three scenarios are possible: (1) a bilinear relationship in which the desorption becomes diffusion limited and thus plateaus, (2) a bilinear relationship in which the adsorption and desorption have different dependences on XSA and (3) a perfect parabola in which the adsorption and desorption are both controlled by XSA.
Chart 1
IV. Further Enantioselectivity Model Development. The Comparative Molecular Field Analysis (CoMFA) model developed in the first year of the grant was very successful in identifying the regions of the molecules in which steric and electrostatic interactions are most correlated with changes in enantioselectivity. However, CoMFA is not very well suited to extrapolate which molecules should be made next. We have developed a new approach that employs simulated annealing algorithms to reduce the number of points in the CoMFA model to identify the most important and then use that simplified model to predict which among a large library of in silico generated catalysts would be the ones to synthesize and test. That concept is illustrated in Scheme 2. The model developed showed excellent regression statistics and allowed for a scan of an in silico generated library of 2705 catalyst structures from which six were chosen for synthesis, all of which showed improved enantioselectivity compared to any of those in the original library, Scheme 3.
Scheme 2
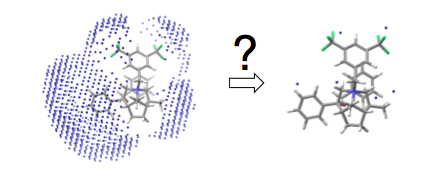
Scheme 3
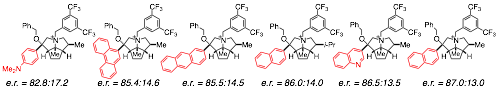
V. Phase Transfer Alkylation of Phenols. The factors that influence the rate of alkylation of the phenol under phase transfer catalysis (PTC) have been investigated (Scheme 4). Six linear, symmetrical, tetraalkylammonium cations: Me4N+, Et4N+, (n-Pr)4N+, (n-Bu)4N+, (n-Hex)4N+, (n-Oct)4N+, were examined to compare the effects of cationic radius and lipophilicity on the rate of alkylation. The tetraalkylammonium phenoxide¥phenol salts were prepared and their intrinsic reactivity was determined from initial alkylation rates with n-butyl bromide in homogeneous solution. The catalytic activity of the same tetraalkylammonium phenoxides was determined under PTC conditions (under an extraction mechanism) employing quaternary ammonium bromide catalysts. In homogeneous solution the range in reactivity was small (6.8-fold) for Me4N+ to (n-Oct)4N+. In contrast, under PTC conditions a larger range in reactivity was observed (663-fold). The effective concentration of the tetraalkylammonium phenoxides in the organic phase was identified as the primary factor influencing catalyst activity. Additionally, titration of active phenoxide in the organic phase confirmed the presence of both phenol and potassium phenoxide aggregates with (n-Bu)4N+, (n-Hex)4N+, and (n-Oct)4N+, each with a unique aggregate stoichiometry. The aggregate stoichiometry did not affect the PTC initial alkylation rates.
Scheme 4