Reports: DNI350395-DNI3: Strategies for the Reduction of Carbon Dioxide to Methanol
 printer friendly
printer friendlyGoal 1: Hydrogen Production using Abundant and Affordable Catalysts
1.1 A Nickel Diphosphine System for Hydrogen Activation and Production
Interest in hydrogen as an alternative fuel stems from its clean-burning properties and the development of fuel cell technology for transportation vehicles. Current limitations to a hydrogen economy are associated with hydrogen storage and hydrogen production, which derives from raw petroleum. There is a strong need for efficient and cost-effective catalysts that can generate hydrogen cheaply on an industrial scale. Platinum is an efficient catalyst for both activating and forming hydrogen; however, it is far from cost-effective. First-row transition metals, which are earth abundant, would be more ideal.
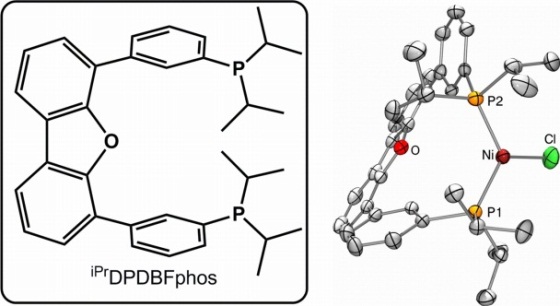
Figure 1. The diphosphine ligand and the Ni(I) chloride complex.
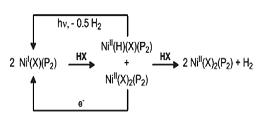
Scheme 1. Possible catalytic scheme for H2 production.
Recently, our lab reported a trigonal nickel(I) chloride complex that is stabilized by the conformationally flexible diphosphine ligand iPrDPDBFphos (Fig. 1). The nickel(I) complex reacts cleanly with vinyl halides in a 2:1 stoichiometry to yield stable Ni(II) products: 2 Ni(X)(iPrDPDBFphos) + CH2CHX → Ni(CHCH2)(X)(iPrDPDBFphos) + Ni(X)2(iPrDPDBFphos). We were intrigued by the possibility of replacing vinyl halides with HX to generate a Ni(II) hydride complex, which could release H2 upon photolysis. These steps would form a similar catalytic cycle for H2 production as shown in Scheme 1. This mechanism is novel because it would only utilize an one-electron redox chemistry. Our nickel(I) halides do react cleanly with HX to form Ni(II) hydride complexes and Ni(II) dihalides. Also, photolysis of the Ni(II) hydride species yields Ni(I) and H2, and this reaction proceeds in the reverse direction, i.e. H2 activation by Ni(I), when heated.
1.2 Synthesis and Characterization of a Nickel Hydride
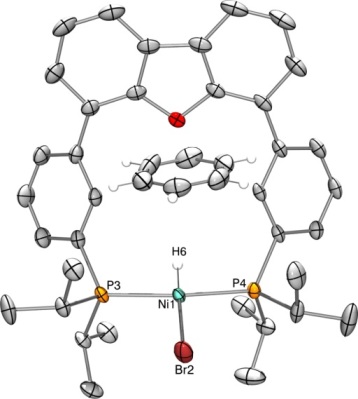
The nickel hydride complexes, Ni(H)(Cl)(iPrDPDBFphos) and Ni(H)(Br)(iPrDPDBFphos) were independently prepared by addition of NaHBEt3 to their dihalide precursors. The hydride signal appears as a triplet at -23.2 and -21.7 ppm (2JHP = 74 Hz) in the proton NMR spectra of the chloride and bromide species, respectively. The Ni-H stretching frequencies are practically identical at 1944 and 1943 cm-1, respectively. A single crystal of Ni(H)(Br)(iPrDPDBFphos) was grown by vapor diffusion of pentane into a saturated benzene solution. An X-ray diffraction study revealed the expected square planar geometry for Ni(II), with the diphosphine ligand chelated in trans fashion. The hydride was located in the difference map, and the Ni-H bond distance was isotropically refined to 1.42(1) �. The structure also reveals one closely packed benzene solvent molecule per nickel complex.
1.3 Reactivity
The Ni(I) chloride reacts with HCl sources to form a clean mixture of Ni(II) hydride and Ni(II) dichloride. The Ni(II) hydride diphosphine complexes release H2 upon photolysis with formation of the Ni(I) halide, and that the complementary reaction, wherein Ni(I) halide activates H2 to form Ni(II) hydride, proceeds to equilibrium. These studies suggest the possibility of a simple hydrogen production scheme that involves only the Ni(II)/Ni(I) redox couple.
Figure 2. X-ray structure of Ni(II) hydride bromide.
Goal 2: Develop New Homogeneous Systems for CO2 Reduction Chemistry
2.1 Metal-Cage Systems.
We developed a novel cage ligand to protect reactive intermediates in a CO2-to-methanol reduction scheme. The cage ligand features a transition metal-binding site located at the bottom of a large cavity, wherein small molecules can diffuse in and become reduced, then the reduced products can flow out. The structure of [PPh4][ZnL] revealed a trigonal monopyramidal coordination geometry for zinc (Fig. 3a).
2.2 Reactivity with Small-Molecules
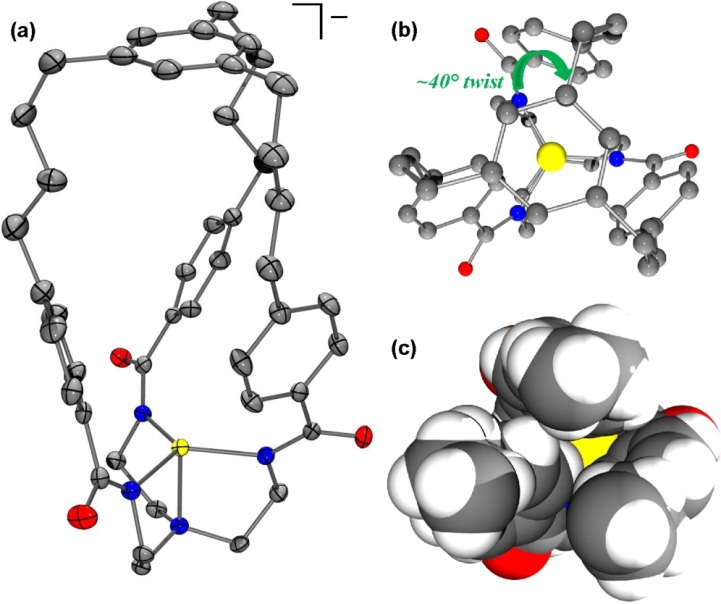
Figure 3. (a) Molecular structure of [PPh4][ZnL]. Top view as (b) ball-and-stick model, or as (c) space-filling model with the aryl cap removed for better visualization of the cavity.
Host-guest chemistry of [PPh4][ZnL] was canvassed with small molecules, e.g. CO2, but the reactions were unproductive. The lack of reactivity is rationalized that the Lewis acidity of the Zn2+ center is significantly attenuated by the ligand's three anionic amide groups.
Goal 3: Develop New Homogeneous Bimetallic Systems for CO2 Reduction Chemistry
In part, due to the cost and time expense associated with making the cage ligands, we have begun to explore using bimetallic systems as new catalysts for CO2 reduction. The creative idea behind using bimetallic catalyst is that we will use a second metal center as a way to tune the activity of the first metal, specifically in the reduction of CO2. This approach contrasts the more common method of modifying the ligand platform, which can be expensive and/or time consuming.
3.1 Heterobimetallics in Small-Molecule Activation
Figure 4. Alternative strategy for heterobimetallic-mediated small-molecule activation
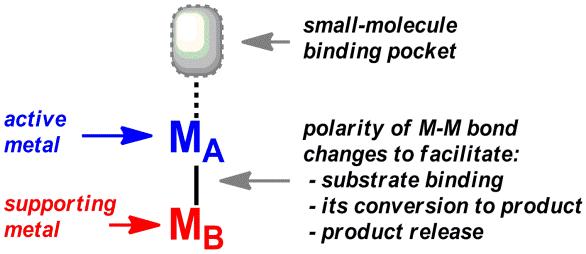
Heterobimetallic chemistry is often touted for its potential to activate small molecules through the cooperative action of Lewis acidic and basic TM sites. The dual "acid-base" character of early-late TM pairs seems especially suited to activate CO2: the basic, late metal attacks the electrophilic carbon center of CO2, while the acidic, early metal stabilizes the nucleophilic oxygen atom. We propose to reduce CO2 using an early-late M-M' bond, as illustrated in Fig. 4. The upper TM (MA) is designated as the active metal because it is the site of small-molecule binding and conversion. The lower TM (MB) dynamically supports the changing electronics at the active metal during each step of the activation process. In this paradigm, the metal-metal bond is central because it is the electronic conduit between the two metal centers.
3.2 Heterobimetallics – Modular Synthesis
The Lu lab has created ligands with a "double-decker" arrangement of donor atoms to form metal-metal (M-M) bonds. These platforms enable the formation of rare M-M bonds in a modular and systematic manner. The versatility of the platforms for making unusual chemical bonds has already been demonstrated by the rapid isolation of three metal-Al(III) (M-Al) bonds (where M is Co, Fe, and Ni), which were exceedingly rare prior to this work (Fig. 5). The Co-Al and Fe-Al complexes are currently being investigated for dinitrogen functionalization.

Fig. 5. Modular construction of M-Al bonds. FeAl and CoAl spontaneously bind dinitrogen (encircled in red).









