Reports: UR449447-UR4: Investigating Proton-Coupled Electron Transfer with Radical Cations Appended with Bases
 printer friendly
printer friendly
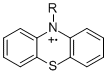 | 2py-PT•+: R = 2-pyridyl 3py-PT•+: R = 3-pyridyl 2pyCH2-PT•+: R = (2-pyridyl)methyl 4pyCH2-PT•+: R = (2-pyridyl)methyl quin-PT•+: R = 8-quinolinyl MeOpy-PT•+: R = 4-methoxyl-3-pyridyl Mepy-PT•+: R = 3-methyl-2-pyridyl |
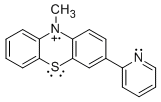 | Me-PT-py•+ |
Student researchers synthesized the precursors for radical cations using procedures modified from the literature, either Buchwald-Hartwig aminations [1] or substitution reactions with the N-deprotonated phenothiazine anion [2]. Cyclic voltammetry affords reversible voltammograms with reduction potentials for B-PT•+/0 ranging from 0.28 to 0.39 V (vs. Cp2Fe+/0 in 0.1 M Bu4NPF6/MeCN). pKa values for +HB-PT were determined through titration with indicators; most were between 10.9-11.9, with +HMeOpy-PT (8.1) and +Hquin-PT (approx. 13.4) as outliers. From these data, effective bond dissociation free energies (BDFEs, for +HB-PT â†' RPT•+ + H•) can be calculated. Most are 78-79 kcal/mol except that for +HMeOpy-PT (74 kcal/mol) [3].
The neutral precursors were oxidized with tris(4-bromophenyl)aminium. Absorption maxima in the UV-vis spectra parallel for other phenothiazine radical cations [4]. All the compounds are stable for hours to days in dry, air-free acetonitrile; the particularly persistent 2-pyPT•+ decayed 2-3% in the absorbance per day. EPR spectra were obtained for 2py-PT•+, 2pyCH2-PT•+ and Me-PT-py•+ in CH2Cl2 in collaboration with Stefan Stoll of the University of Washington. They suggest that the unpaired spin density is predominantly on the phenothiazine ring with negligible density on the pyridine ring. All the reactivity studies were performed with radical cations generated in situ in this manner.
Phenothiazine radical cations can accept an electron, and pyridine can accept a proton. The combination of these two components will allow for the net transfer of a hydrogen atom by CPET. Hence, we explored the reactivity of these radical cations with two hydrogen atom donors, 2,4,6-tri-tert-butylphenol and 2,6-di-tert-butyl-4-methoxyphenol with stopped-flow UV-vis.
Addition of the phenols to 2py-PT•+, 3py-PT•+, 2pyCH2-PT•+ and Me-PT-py•+ in acetonitrile leads to the decay of the absorptions in the visible region of the spectrum. In reaction with 2,4,6-tri-tert-butylphenol, absorptions corresponding to the phenoxyl radical appear. Kinetic studies monitoring the decay of the radical cations in excess phenol provided the expected pseudo-first-order decays, and the slopes of the pseudo-first-order rate constants vs. concentrations of phenol plots provide second-order rate constants. Isotope effects were determined for 2py-PT•+, 2pyCH2-PT•+ and Me-PT-py•+ that ranged from 2.6-8.7, and in addition, rate constants for all four compounds with 2,6-di-tert-butyl-4-methoxyphenol were determined.
With some additional data, the kinetic and thermodynamic data support the CPET. Bimolecular kinetics support both the radical cation and the phenol in the rate-limiting mechanism, and the isotope effect supports the involvement of the proton. Using literature values for the phenols [3] the ΔG° for initial electron transfer can be directly calculated with experimental reduction potentials for B-PT•+/0, and the ΔG° for initial proton transfer can be determined using the reduction potential for HB-PT•2+/+ (by cyclic voltammetry on the protonated compound) and a thermodynamic cycle. In all cases, the experimental barrier ΔG‰ (calculated from the rate constants) for reaction is smaller than that the ΔG° for reaction to form potential intermediates.
Computational studies by Jeffrey Wolbach and his student co-workers also suggest that these reactions occur by a CPET mechanism. Geometries were calculated with B972/6-31+g(d,p) with thermal corrections. The reactants, transition states (TSs), and products for CPET mechanistic step for reaction between several radical cations and the phenols have been calculated. (The reactions occur between hydrogen-bonded complexes.) The TSs were verified both with imaginary frequency and intrinsic reaction coordinate calculations. While the proton transfer intermediates (HB-PT•2+ + ArO--) are significantly higher than the TS energy, the electron transfer intermediates (B-PT + ArOH•+) are close to the CPET TS energy. The transformation of 2py-PT•+ to +H2py-PT occurs with a significant conformational change, from a planar phenothiazine ring with an orthogonal pyridine to a bent phenothiazine ring (the N is pyramidalized) with a quasi-equatorial pyridine.
In the next year, we intend to complete this series and investigate new compounds to determine how changing geometry and electronics affect the mechanism and BDFEs.
References
[1] (a) Driver, M. S.; Hartwig, J. F. J. Am. Chem. Soc. 1996, 118, 7217-7218. (b) Franz, A. W.; Popa, L. N.; Frank Rominger, F.; Müller, T. J. J. Org. Biomol. Chem. 2009, 7, 469-475.
[2] Chen, P.; Westmoreland, T. D.; Danielson, E.; Schanze, K. S.; Anthon, D.; Neveux, P. E., Jr.; Meyer, T. J. Inorg. Chem. 1987, 26, 1116-1126.
[3] Warren J. J.; Tronic, T. A; Mayer, J. M. Chem. Rev. 2010, 110, 6961-7001.
[4] Rosokha, S. V.; Kochi, J. K. J. Am. Chem. Soc. 2007, 129, 3683-3697.









