Reports: ND150715-ND1: Organocatalytic Tether Formation
 printer friendly
printer friendlyIntroduction
Catalysis of intermolecular reactions is at the basis of chemical reactivity. Bifunctional catalysts and enzymes are particularly effective, as they perform substrate activation while favoring substrate preassociation. Typically, synthetic catalysts attempt to emulate the remarkable efficiency of enzymes in catalyzing intermolecular reactions (with rate accelerations as high as 1017). To achieve high activity and control, enzymes pre-organize reagents and consequently induce a "temporary intramolecularity" that minimizes the entropic penalty associated with intermolecular reactions. It is accepted that rate accelerations of 104-108 for 1 M reactants can be obtained for room temperature reactions via temporary intramolecularity. Not surprisingly, multiple transformations building on preassociation have been developed, and several families of bifunctional catalysts have emerged. In contrast, the catalysis of transformations only through temporary intramolecularity has received little attention from the synthetic community. Indeed, simple catalysts operating only through this pathway are rare, perform relatively simple reactions and highly efficient asymmetric catalysts have not been reported.
In late 2011, our group reported the use of simple aldehydes as tethering catalysts. Building on Knight's seminal work, we showed that selected aldehydes catalyze intermolecular Cope-type hydroaminations via temporary intramolecularity. As illustrated below, the reaction proceeds via in situ formation of a mixed aminal (I), which allows for a facile intramolecular hydroamination event. As opposed to traditional tethering strategies, this catalytic method does not require additional synthetic steps for the installation and cleavage of the tether. Furthermore, enantioenriched molecules can be formed using chiral aldehydes, which efficiently transfer stereochemical information. Following our initial communication we have gained solid mechanistic understanding, which includes the rate law of the reaction and identification of two catalyst inhibition pathways. This work also showed why catalysts possessing a destabilized ground state are more reactive: they favor the required preassociation event, and this Keq is in the rate law of the reaction. This understanding led to the identification of an improved achiral catalyst, formaldehyde, which is more active and allows (significantly) more difficult hydroaminations to proceed. In addition, we have identified an optimal bicyclic chiral catalyst, which is remarkably robust toward epimerization and achieves high enantioinduction (94%ee).

Progress report
The purpose of this Grant is to expand this catalytic tethering approach to other reactions than Cope-type hydroamination. The goals of the original proposal fell into three categories: catalyst optimization, reaction discovery and enantioselective catalysis. The first objective is to provide an in-situ, catalytic alternative to known tethered strategies. The second objective is the development of novel reactivity, including reactions relying on concurrent tandem catalysis. The third and final objective is to explore stereoselective variants using a chiral organocatalyst. As described below, solid progress has been made toward these objectives.
In early 2011, exploration began toward novel reactivity. Initial attempts were focusing on directed Huisgen cycloadditions (click reaction, Eq 1) for several reasons. Notably we felt that reports of thermal, intramolecular reactions occurring at 50-100°C were encouraging. This system also appeared optimal to explore concurrent tandem catalysis with transition metal complexes, possibly resulting in milder conditions. Unfortunately, catalytic reactivity could not be achieved despite considerable experimentation, including the evaluation of each step of the putative catalytic cycle. Overall, these early efforts suggested that more understanding of the basic reactivity was required, and led us to investigate the mechanism of the aldehyde-catalyzed hydroaminations in parallel to the exploration of novel reactivity.
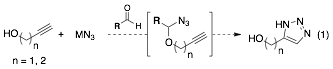
Starting in the fall of 2011, we pursued the development of two other reactions: mono-alkylation of amines (see proposal) and carbonyl-catalyzed hydrolysis reactions. Efforts toward mono-alkylation of amines were exploratory and highlighted the existence of background (uncatalyzed) reactivity under some of the reaction conditions screened (Eq 2). In parallel, we reinvestigated a hydrolysis system pioneered by Commeyras to gain solid footing toward new reactivity. An illustrative example is the formaldehyde "catalyzed" hydrolysis of α-aminoamides (Eq 3).
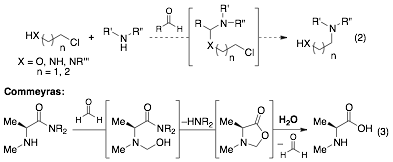
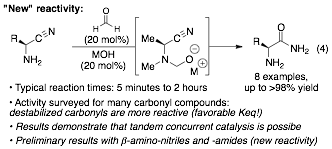
Over 25 years ago, Commeyras developed various carbonyl directed hydrolysis reactions via hemiaminal intermediates. Carbonyl-catalyzed α-amino-ester, -amide, -thioamide and -nitrile hydrolyses were reported. However, these reactions used stoichiometric amounts of a carbonyl catalyst, even though turnover should occur. In addition, the highest enantioselectivity reported is 42%ee. This work is important from a tethering standpoint even if the "tethered nucleophile" can only be water and if the issue of catalytic activity (turnover frequency and rate) has not been addressed. Our efforts in this system have proved rewarding: 1) a version using catalytic amounts of the carbonyl compound and of the base has been developed (Eq 4; constituting proof of concept results on the concurrent tandem catalysis objective); 2) reactivity with various carbonyl catalysts mirrors what is observed for hydroamination, indicating that destabilized aldehyde catalysts are optimal to promote preassociation (favorable pre-equilibrium); 3) formaldehyde is an optimal catalyst for the transformation (i.e. a more efficient "tethering" system has been identified); 4) this works provides relatively simple reactivity to explore enantioselective variants of these reactions (just started!).
Important findings secured during the hydroamination studies are that highly effective asymmetric catalysis is possible, and that the stereochemical integrity of bicyclic aldehyde catalyst is excellent under basic conditions. The need to achieve kinetic control during the preassociation event remains a challenge, since thermodynamic control should result in a poor selectivity during the formation of a chiral tether and, consequently, to poor enantioselectivity.
Future Plans and Outlook
The results achieved to date and the financial resources remaining show that we are well positioned for progress in the coming year. Efforts toward the first objective will involve optimized catalysts (e.g. formaldehyde) to show that "transiently" tethered cycloadditions ([4+2] and [2+2]) are possible. Development of new reactivity will involve a-aminoacid derivatives, and will be expanded to b-aminoacid analogues (encouraging results have been obtained). Exploration of reactions featuring concurrent tandem catalysis with transition metals will be continued. Finally, the a-aminoacid system appears optimal for further development of asymmetric tethering organocatalysis. We believe that a first communication on new reactivity via "organocatalytic tether formation" is likely for 2013 and that the results on current objectives will indeed highlight the synthetic potential of catalytic approaches relying only on temporary intramolecularity.









