Reports: ND449785-ND4: Topologically Diverse New Polycyclic Scaffolds via Key Photochemical Steps
 printer friendly
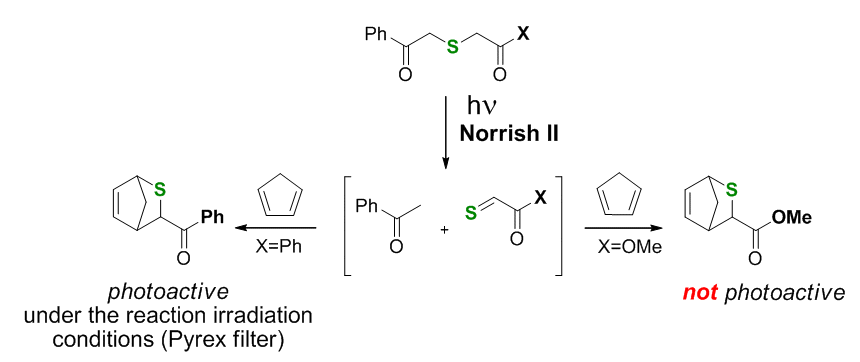
printer friendlyThe proposed research is to develop a series of efficient synthetic pathways to topologically diverse polycyclic and polyheterocyclic scaffolds by combining rationally selected thermal and photoinduced cycloaddition reactions. In the last year of the proposal we have developed a one pot photoinduced tandem transformation, incorporating the generation of thiocarbonyl compounds via a Norrish Type II fragmentation in phenacyl sulfides. Vedejs (1982) showed that phenacylsulfides (Scheme 1) were readily prepared and irradiated to generate thioaldehydes which underwent [4+2] cycloaddition with cyclopentadiene to give respective thianorbornenes. This worked well for the shown methoxycarbonyl derivative (X=OMe, right). However, the method was not ideal for photogeneration and cycloadditions of thiacarbonyl compounds conjugated to benzoyl (X=Ph, left), because the same chromophore, the benzoyl group, was incorporated in the resulting Diels-Alder adduct which, the authors caution, is "destroyed by prolonged irradiation."
Scheme 1.
We were wondering if this reported "destruction" upon extended irradiation can be, in fact, a useful transformation that the authors simply overlooked. In the course of our studies we probed this hypothesis by (i) generating the Diels-Alder adduct of phenyl(thio)glyoxal via non-photochemical means and examining photochemistry of the adduct and (ii) incorporating the two photochemical steps into a single cascade and carrying out a "one-pot" photochemical generation of the thioaldehyde, its[4+2] cycloaddition, and subsequent photo-rearrangement of the [4+2] cycloadduct – all starting from bis(phenacyl)sulfide.
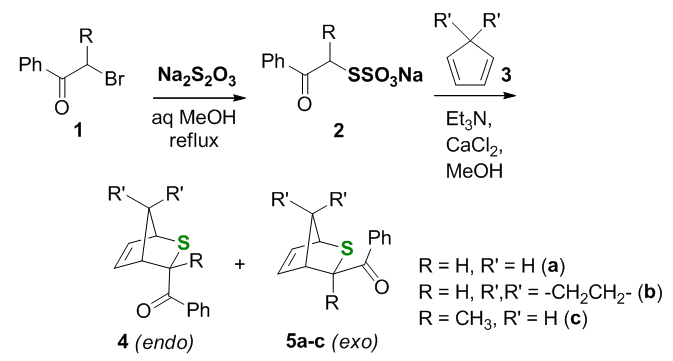
Our first objective was to generate a potentially photoactive [4+2] cycloadduct of a thioaldehyde and a diene via a non-photochemical route, to avoid premature secondary phototransformation of the cycloadduct. For this reason we chose Kirby's Bunte salts-based method for generation of phenyl(thio)glyoxal, which was produced by base-catalyzed sulfite elimination in the presence of cyclopentadiene or sprio-2,4-heptadiene to yield thiabicyclo/tricyclo alkenes, possessing photoactive benzoyl pendant, as shown in Scheme 2.
Scheme 2.
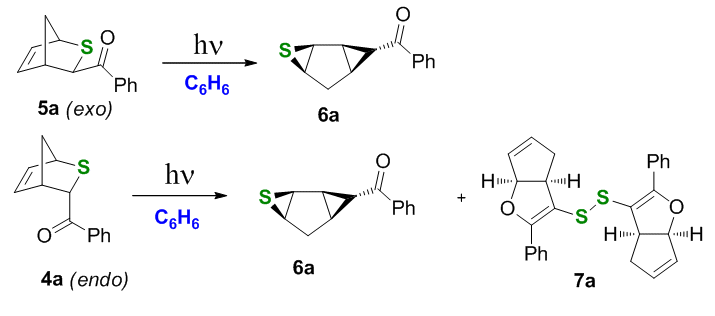
It is easy to see that only one photoreactivity mode is available to the exo-thianorbornenes 5, that is the C-S bond fragmentation because the a-sulfide bridgehead hydrogen is not accessible for abstraction and the double bond is not accessible for the exo-benzoyl group either. The excited carbonyl group in the endo-isomers 4, while also can expel the sulfanyl radical, has another option – to attack the double bond as in the Paternò-Büchi cycloaddition.
Irradiation experiments in benzene with a UV source confirmed this hypothesis. Upon irradiation in benzene exo-photoprecursor 5a yielded tricyclic thiirane 6 nearly quantitatively, whereas the endo-precursor 4a exhibited dual photoreactivity, furnishing both thiirane 6a and disulfide 7a.
Scheme 3.
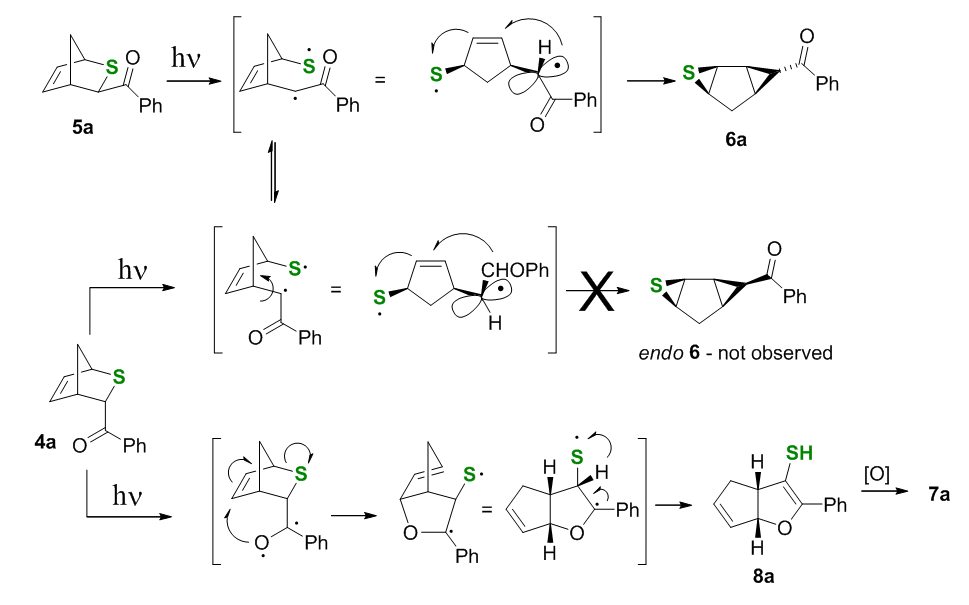
Our rationale for the formation of thiirane 6a involves photoinduced BzC-S homolytic bond fragmentation followed by radical addition to the endo-cyclic double bond, Scheme 4, which probably occurs stepwise or, at least, asynchronously. Before the phenacyl radical reacts it apparently has time to rotate, so both the endo and the exo isomers produce the same exo-Bzthiirane 6a.
The mechanistic rationale for the oxapentalene channel starting from endo-isomer 4a involves an interrupted Paternò-Büchi reaction, where the expulsion of the sulfanyl radical from the initially generated 1,4-diradical occurs faster than radical recombination forming the oxetane ring. Subsequent hydrogen migration, which amounts to disproportionation in the 1,3-diradical yields the thiol and the disulfide. The disulfide is formed in the presence of air, which is common for photochemical reactions of thiols.
Scheme 4.
Thioketones also undergo [4+2] cycloaddition to cyclopentadiene. For example, the diene's reaction with 1-phenyl-2-thioxo-propan-1-one produces a mixture of exo (5b) and endo (4b) stereoisomers, which were irradiated under the same conditions as above.
Scheme 5.
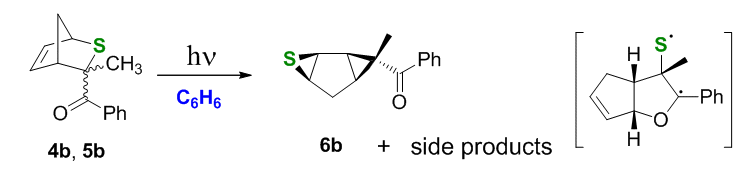
In this reaction thiirane 6b was the only dominating product, with no oxapentalenederivatives observed (Scheme 5). This can be explained by both (i) a weaker aC-S bond (expected to fragment much faster) and/or (ii) a methyl-induced less favorable conformation of the benzoyl group in the endo-isomer which slows the interrupted Paternò-Büchi channel.
Spiro-photo precursors 4c and 5c were obtained by reacting phenyl(thio)glyoxal, generated from the Bunte salt 2,with spiro-heptadiene as shown in Scheme 2. As it is abundantly clear from Scheme 6, their photoreactivity is similar to that of thianorbornenes 4a and 5a.
Scheme 6.

It appears that the interrupted Paternò-Büchi reaction in endo-3-benzoyl-2-thianorbornenes is general.
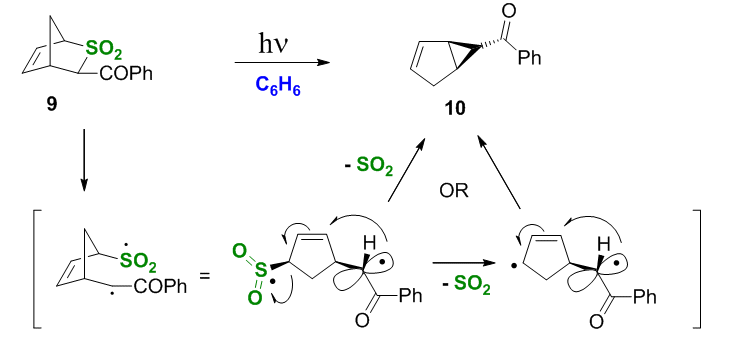
We also explored the photochemistry of the corresponding sulfones, which were obtained by oxidation of the thianorbornenes 4a and 5a with mCPBA. Regrettably, the acidity of the C(3)-H proton prevented us from obtaining the endo-Bz epimer due to enolization and rapid conversion of the endo isomer into the exo. The exo isomer 9 undergoes the aC-SO2 bond scission with subsequent extrusion of SO2 either (i) assisted by the phenacyl radical via the allylic radical substitution or (ii) a two-step dissociative mechanism, Scheme 7.
Scheme 7.
After exploring the photochemistry of adducts 4 and5 prepared via the ground state chemistry, i.e. non-photochemically, we explored the feasibility of a one-pot integrated procedure for photogeneration of phenyl(thio)glyoxal from phenacyl sulfide, its [4+2] cycloaddition to cyclopentadiene, and subsequent photoinduced transformation of thianorbornene equipped with the photoactive benzoyl group.
Scheme 8.
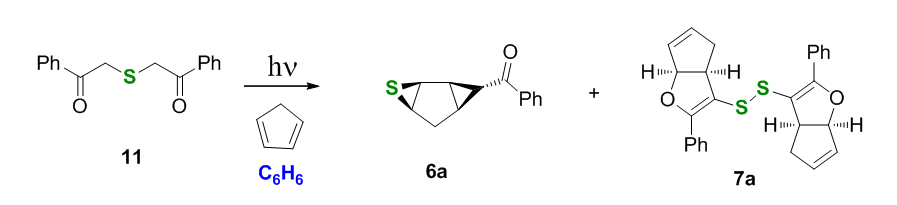
We found that the kinetics of the ground state phenyl(thio)glyoxal addition to cyclopentadiene is a limiting factor. Excess of cyclopentadiene helps capture the photogenerated thiocarbonyl. In stoichiometric runs the photogenerated phenyl(thio)glyoxal undergoes secondary transformations. It was further found that solvent had a dramatic effect on the formation of disulfide 7a. For example, running the reaction in dichloromethane or chloroform produced thiirane 6a as the major product, but no disulfide 7a was formed. Instead, a completely desulfurized oxapentalene 12a was detected in a 2:3 ratio to thiirane 6a.
Scheme 9.

In summary, we have investigated a new photoinduced transformation of thianorbornenes outfitted with a photoactive benozyl pendants. The primary photochemical product is thiirane 6, which is derived from homolytic scission of the BzC-S bond upon excitation of the benzoyl chromophore. We have also discovered a novel interrupted Paternò-Büchi channel in these irradiations leading to oxapentalene structures. While the yield of this photochemical transformation is modest (20%, calculated based on the convertedphenacylsulfide 11), it gives rapid access to fused dihydrofuran derivatives via a one pot photoassisted reaction starting from readily available materials.









