Reports: DNI350843-DNI3: Trinuclear Copper Complexes for Dioxygen Activation and Reduction
 printer friendly
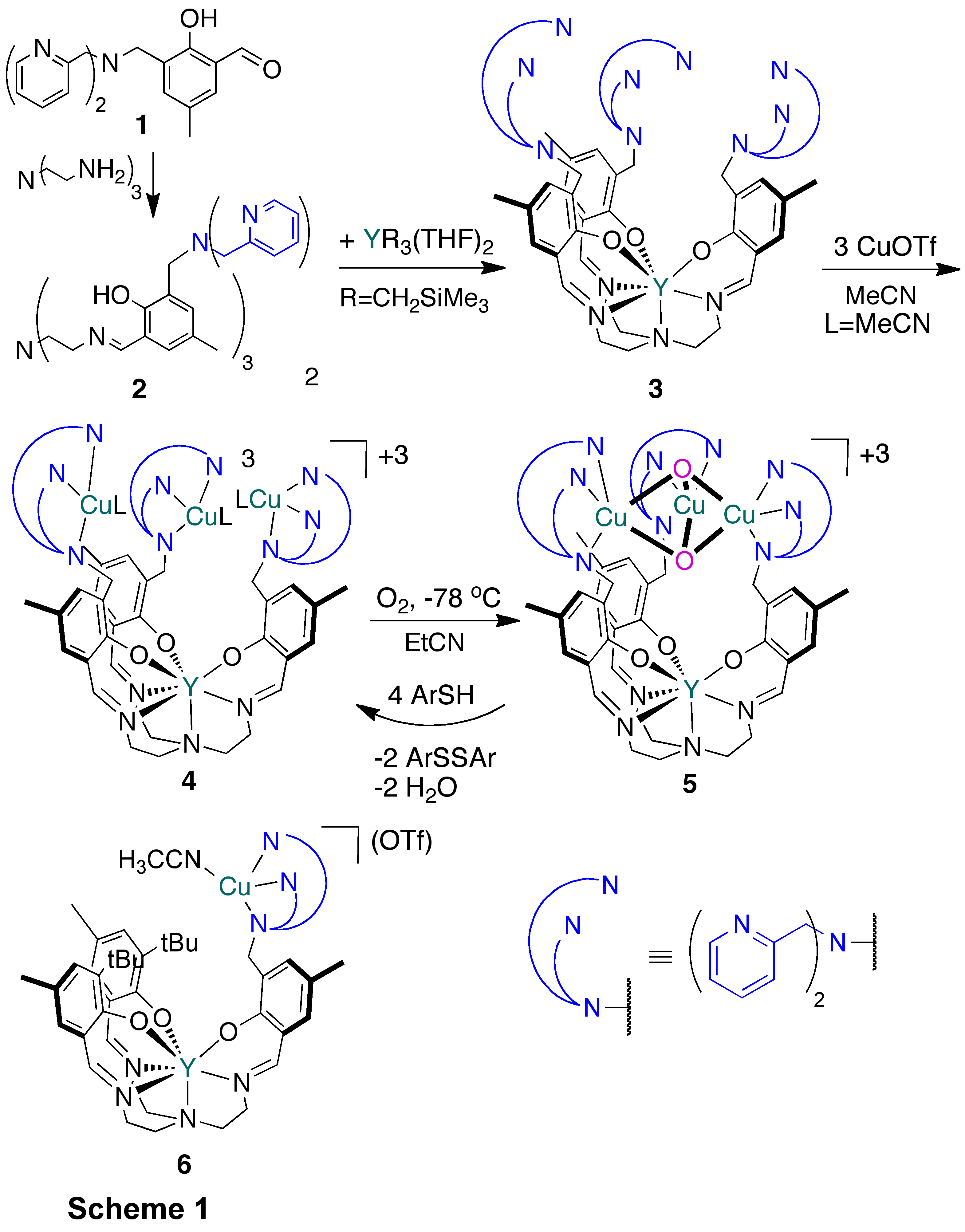
printer friendlyEnzyme active sites containing three metal centers perform diverse biological functions, including electron transfer, hydrolytic reactions, and O2reduction. In the context of O2 reduction to water, the tricopperactive sites of laccase and ascorbate oxidase (AO) have been studied extensively given the potential applications in fuel cell chemistry. Synthetic efforts have focused on modeling the tricopper site where O2activation occurs. Mononuclear diamine complexes have been shown to self-assemble upon reaction with O2 to form a CuII2CuIII(m3-O)2species. Reduction of these trinuclear cores leads to dinuclear species,however, hindering attempts for developing catalytic versions of this reaction. An alternate strategy has been the employment of trinucleating frameworks. Most systems investigated are rather flexible and do not faciliate cooperative trinuclear reactivity. We have developed a novel strategy for the generation of trimetallic moieties based on templating a ligand scaffold around yttrium orlanthanides to generate a seven-coordinate complex that displays three multidentate sites for binding of transition metals.
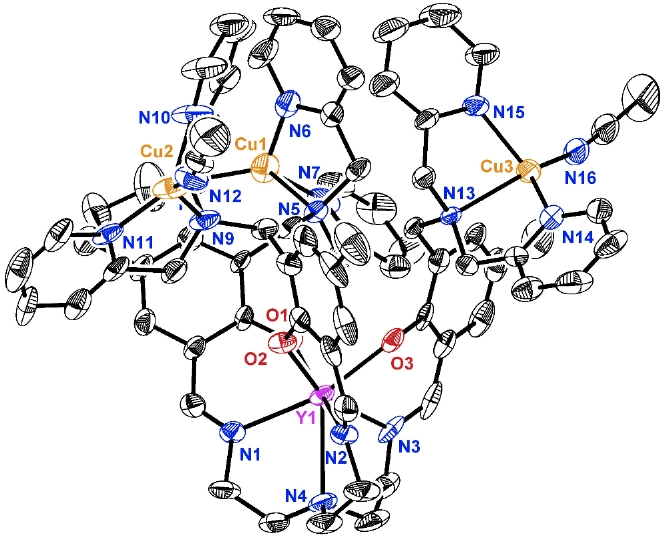
Toward assembling a multinucleating ligand architecture, a variant (2) of the [O3N4]-trisphenoxide-trisimine-amineframework with dipicolylamine substitutents was prepared via a condensation between tris(2-amino-ethyl)amine and three equivalents of previously reported aldehyde 1(Scheme 1).Metallation was perfomed via alkane elimination with an yttrium trialkylprecursor. NMR spectroscopic characterization shows a single phenoxide andpyridine environment, consistent with yttrium binding to the [O3N4]-trisphenoxide-trisimine-aminemoiety to generate a high-symmetry species (3). Addition of three equivalentsof CuI precursor (Cu(CH3CN)4OTf) dissolved in acetonitrile (MeCN) to a suspension of 3 in MeCN leads to the generation of a homogeneous mixture and to a color change from light yellow to gold. Single crystal X-raydiffraction studies show the formation of a tricationic Cu3Y species(Figure 1), with the seven-coordinate yttrium center bound to the [O3N4]-trisphenoxide-trisimine-aminesite, the three copper centers bound to the dipycolylamine moieties, and three outer-sphere triflate anions. The selectivity for yttrium binding to the [O3N4]motif, despite the presence of nine additional nitrogen donors is notable, and indicates that this strategy may be more general with respect to changes in the nature of chelating ligands and metals decorating the [YO3N4]unit.
The reaction of compound 4 with O2 was monitored by UV-Vis spectroscopy. The new species (5) generated is stable for at least 24 hours at -78 °C. Volumetric measurements carried out with a Toepler pump indicate that 0.97±0.01 equivalents of O2 are consumed per equivalent of 4, supporting the formation of a complex with the stoichiometry "Cu3O2". This corresponds to the CuII2CuIII(m3-O)2 core previously reported with bidentate ligands: N,N,N',N'-tetraalkylcyclohexanediamines(A, for N,N,N',N'-tetramethylcyclohexanediamine;Stack), N,N-dimethyl-2-picolylethanamine(B, Itoh), and more recently 4-(2,2-dimethylhydrazino)-dimethylhydrazone-3-penten-2-one (C,Tolman). These complexes were all self-assembled from concentrated solutions of monocopper(I) precursors supported by bidentate amines. Spectroscopic comparison with A, B, and Cand the reaction stoichiometry support the assignment of species 5 as displaying the CuII2CuIII(m3-O)2 motif.
The synthesis of 5is notable given the lack of precedent for preassembled trinuclear complexes that show similar reactivity with O2. The generation of 5 is particularly remarkable given that previous studies of copper-dipicolylamine complexes do not report trinuclear reactivity. For comparison, a related monocopper complexes was prepared, 6, displaying the copper-dipicolylaminemoietiy. Complex 6 is analogous to 4 by containing the Y-[O3N4] motif, but displays a single dipicolylamine arm resulting in amonocopper complex. If intermolecular copper self-assembly reactivity upon reaction with O2 was responsible for the spectroscopic features of 5, mononuclear CuI complex 6 were expected to undergo reactivity similar to 4 given the steric and electronic similarities. Reaction of either complex with O2, however, leads to a species that has spectroscopic features different from 5, even at high concentrations. These findings suggest that the reactivity of precursor 4 is intramolecular in copper and, moreover, the formation of the CuII2CuIII(m3-O)2 moiety is facilitated by the presence of the three Cu centers in close proximityon the same ligand framework.
The reactivity of complex 5 with reducing agents was tested for regeneration of Cu(I) precursor. Upon treatment with 4-tert-butylthiophenol disappearance of the UV-Vis bands characteristic of 5 was observed within 1 min at -40°C. After anaerobic workup at room temperature, the reaction mixture contained two equivalents of the diaryldisulfide product, corresponding to an overall transfer of four protons and four reducing equivalents by complex5. A reduced (diamagnetic) tricopper species was recovered in near-quantitative yield and was found to displaythiophenol ligands. The identity of the recovered tricopper complex (4⋅3ArSH) was confirmed by independent synthesis via addition of thiophenol to 4. The present Cu3(m3-O)2 moiety is a weaker oxidant relative to other dicopper and tricopper oxo species with reported reduction potentials. This may explain its lack of oxidative C–C coupling reactivity with alkylphenols. This behavior is consistent with the function of tricopper sites in laccases and AOs, which are oxidases and hence do not perform C–H oxygenations.
In summary, ligand frameworks displaying a total of sixteen or thirteen oxygen and nitrogen donors was selectively templated byyttrium(III) binding to a tripodal [O3N4]-trisphenoxide-trisimine-aminemoiety. The remaining three pycolylamine-based metal binding sites coordinate copper(I). Reactions of these CuI3Y complexes with O2generate CuII2CuIII(m3-O)2 motifs according to spectroscopic comparisons and the measured reaction stoichiometry. Oxidation of thiol to generate two equivalents of diaryldisulfide show the ability of 5 to reduce to a tricopper(I) core using four electrons and four protons. Starting from 4, the overall process corresponds to the reduction of O2 to H2O. Future studies will focus in developing catalytic O2 reduction systems and mechanistic studies of O2activation. The reported molecular architectures are expected to be generally applicable to the assembly of multimetallic complexes for transformations including multielectron redox processes and acid-base chemistry. The ACS PRF funding has been instrumental in starting this project. A talented graduate student and a minority undergraduate have been funded on this grant. The graduate student is continuing his work on projects involving O2chemistry. The undergraduate is planning to pursue graduate studies in chemistry and is applying for admission this Fall. A manuscript describing thiswork is in preparation.