Reports: UNI350980-UNI3: Ambiphilic Scaffolds for Cooperative Metal-Ligand Activation of Small Molecules
 printer friendly
printer friendlyThe goal of our PRF-supported research, and an overarching theme of my laboratory, is the construction and elucidation complexes containing a relatively electropositive element multiply bonded to a late transition metal. As described in a recent publication from my laboratory, this type of bonding situation may lead to complexes that serve as metal-based analogues of frustrated Lewis pairs (FLPs), where the Lewis acidity of the electropositive ligand and the Lewis basicity of the metal remain unquenched and lead to novel reactivity. In particular, we have focused on pincer-type complexes where the reactive electropositive element (silicon, boron, or carbon) is stabilized by tethering to the metal through strong phosphine donors (one formulation is depicted in Figure 1).
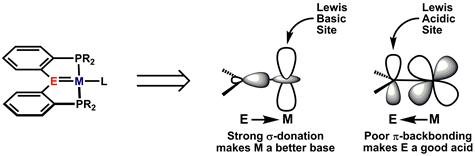
Figure 1. Pincer-type complexes containing adjacent Lewis acidic and basic sites (E = Si, B, C, inter alia)
The first goal of this project was to develop reliable syntheses for ligand precursors that could be incorporated into such a complex. We chose [RP2Si]H2 ligands with phenylene backbones (e.g., complex 1 in Scheme 1 as a set of initial targets due to the ready accessibility of o-brominated phenyphosphines through established cross-coupling methods and previous reports indicating that such complexes are useful precursors for multidentate phosphinosilyl ligands. We pursued parallel routes to the synthesis of the diaryl dihydrosilanes: (1) substitution at SiCl4 followed by reduction with lithium aluminum hydride (Scheme 1, top), and (2) substitution at the H2SiCl2 surrogate, (teeda)SiH2Cl2 (Scheme 1, bottom). Both routes have been effective for synthesis of the ligand precursor.

Scheme 1. Two demonstrated routes to synthesis of [RP2Si]H2 ligand precursor 1 (R = Ph, Cy)
The top route (substitution then reduction) has the advantage of being less costly and not requiring the synthesis of the starting silicon electrophile, but we have shown that oversubstitution at silicon can be a problem for ligands with less bulky phosphine substituents. The bottom route (substitution at a pre-reduced silicon center) has the advantage of preventing oversubstitution and allowing the possibility of a one-step preparation, but has the disadvantage of requiring synthesis of the masked H2SiCl2 unit and requiring separation of tetraethylethylenediamine (teeda) from the product mixture. Ultimately, it seems that each method will find its niche for a particular synthesis. We have been particularly pleased with how well the (teeda)SiH2Cl2 synthesis has proceeded, since few reports of the successful use of this reagent are present in the literature, and we certainly plan to publish this route to demonstrate the potential utility of (teeda)SiH2Cl2 as a dichlorosilane surrogate.
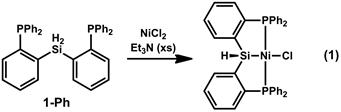
Preliminary studies of the coordination chemistry of 1 have shown that it will cleanly form complexes with nickel, cobalt, and rhodium, though the rhodium complexes seem to form as a mixture of several closely related isomers that have not been definitively identified. In the case of nickel, metallation in the presence of sacrificial base (Et3N) appears to lead to elimination of HCl and formation of the diamagnetic four-coordinate (PhP2SiH)Ni–Cl (Eq 1). Reactivity studies with these complexes have just begun and will include attempts to form metal silylenes both by reduction with hydride sources (to induce elimination of H2) and hydride abstraction using Lewis acids. Upon obtaining the desired metal silylene complexes, reactivity with both nonpolar (e.g., C–H) and polar (e.g., C=O) multiple bonds will be explored with the goal of developing catalysis using the silylene (or hydrosilyl) as a hydride shuttle in reductive or oxidative catalysis with a particular focus on petroleum feedstocks. Additionally, preliminary results indicate that the cyclohexyl-substituted ligand will be a more useful supporting ligand for metals such as rhodium and iridium that are prone to cyclometallation, and we look forward to exploring this newly prepared ligand in our second year of PRF funding.
In addition to the results presented above, we have recently begun the preparation of pincer-type N-heterocyclic silylene ligands for comparision with the aryl-substituted versions already in hand. The proposed synthesis is shown in Scheme 2. Thus far, we have been able to cleanly obtain large amounts of the intermediate prior to silicon-atom incorporation, and we will focus increasing efforts on the final step over the coming months. To the best of our knowledge, N-heterocyclic silylene pincers have not been prepared and would provide an excellent comparison with the more acidic silylenes targeted to this point.
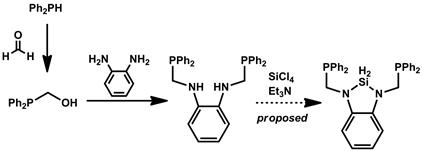
Scheme 2. Partially completed synthesis of a new N-heterocyclic silylene pincer ligand
Support from the ACS-PRF during the first year of the project described above has funded the research of two undergraduate students and has allowed us to make substantial progress toward the preparation of new ambiphilic scaffolds that contain acidic main-group elements multiply bonded to basic late transition metals. Preliminary insights into this and related work have been published and both students will be presenting their results at professional meetings during the coming year. The next grant year will be spent focusing our efforts on the successful results obtained thus far in order to begin determining the types of reactivity that can be educed from this new class of metal complexes.









