Reports: UNI150667-UNI1: Synthesis and Borate-Catalyzed Kinetic Resolution of Terminal Aziridines
 printer friendly
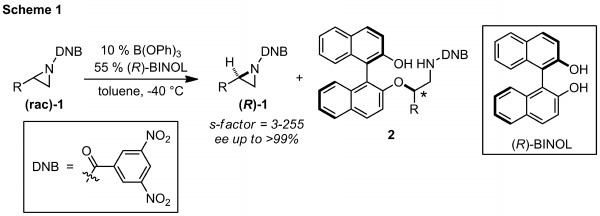
printer friendlyThe ACS Petroleum Research Fund has supported our groups efforts to developed a highly selective kinetic resolution of N-acylaziridines. In this process, racemic aziridines (1), available readily from alkenes, are treated with enantiopure 1,1'-bi(2-naphthol) ((R)-BINOL) catalyzed by triphenylborate (B(OPh)3) to facilitate enantioselective addition leading to kinetic resolution (Scheme 1). A 3,5-dinitrobenzoyl (DNB) nitrogen protecting group was vital for successful resolution. The reaction is generally selective for terminal DNB aziridines where the R group is a primary or secondary carbon. If the R group is too small (methyl) or too large (tert-butyl), selectivity drops dramatically. Successful resolutions were also completed with cis-disubstituted DNB aziridines, where trans-disubstituted aziridines were poor substrates. The consumed enantiomer of aziridine undergoes ring-opening to produce byproduct 2 in which nucleophilic attack had occurred on the more hindered carbon for terminal aziridines. The observed selectivity, attack at the more hindered carbon, intrigued us since literature examples strongly suggest the opposite regioselective for most aziridine opening reactions, even under Lewis acid conditions. A full account of substrates and reactivity of 2 can be found in our recent publication (DOI: 10.1002/anie.201204224). Extension to other nucleophiles was pursued with enantioenriched aziridine obtained from a gram scale kinetic resolution.
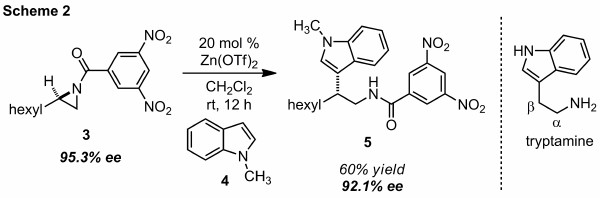
Initially, carbon nucleophiles were investigated under Lewis acid conditions generating a new carbon–carbon bond attached to a stereogenic carbon. We demonstrated that zinc triflate is a competent catalyst for the addition of N-methylindole (4, 4 equiv) to N-acylaziridine 3 in 60% yield with only a slight loss of enantiopurity (Scheme 2). The preference for internal aziridine attack was confirmed by COSY and HMBC studies on tryptamine 5. The beta-substituted tryptamine product produced from this route are structurally unique, since only alpha-substituted tryptamines are prevalent in nature.
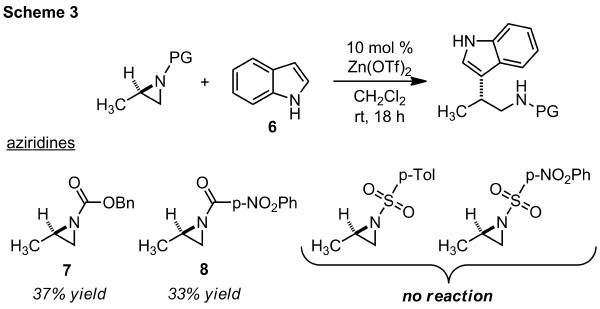
The 3,5-dinitrobenzoyl (DNB)-protected aziridines performed best in our kinetic resolution and was therefore chosen for identification of carbon nucleophiles. We have been working with the DNB protecting group for several years and while some DNB-protected aziridines undergo rapid rearrangement, no violent reactivity has been observed even under heating conditions. The DNB protecting group can be removed following aziridine ring opening; however, we considered other aziridine protecting groups could perform better. We have demonstrated that Cbz- and p-nitrobenzoyl-protected aziridines (7 and 8) undergo nucleophilic ring opening with indole (6), but with lower yields that the DNB-protected aziridine (Scheme 3). In both cases, the major isolated product resulted from stereospecific attack at the more hindered carbon. Interestingly, sulfonyl aziridines were inert to the zinc-catalyzed conditions (Scheme 3). These data suggest that DNB-protected aziridines are uniquely suited for regioselective nucleophilic ring opening at C2.
The moderate yield observed under our initial zinc-catalyzed indole addition reaction conditions is partially linked to competitive rearrangement of N-acylaziridine (9) to oxazoline (11, entry 1, Table 1). However, with only a 57% mass balance, other decomposition must be occurring despite the use of excess 6. A screen of Lewis acids was performed using an NMR internal standard to determine product yield. A series of triflate salts produced similar amounts of addition and oxazoline (entries 2–5). Lanthanum triflate was initially promising with a high 10:11 ratio, but longer reaction times and heating failed to increase the yield of 10 beyond other triflate salts (entry 4). Our hope was to develop a process employing catalytic amounts of Lewis acid; however, the lack of success identifying an improved metal-triflate salt led us to consider a stoichiometric Lewis acid known to facilitate aziridine opening. Indeed, boron trifluoride (BF3) was the superior Lewis acid promoting ring opening at -78 °C in one hour. The reaction produced the highest yield to date with complete stereochemical transfer to tryptamine 10 and no oxazoline observed. With optimized conditions in place, we are poised to develop the indole addition further.

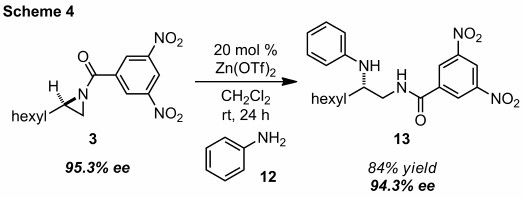
Access to large quantities of enantioenriched terminal DNB-protected N-acylaziridines from our kinetic resolution studies prompted us to investigate heteroatomic nucleophiles for stereospecific ring opening. Employing the initial zinc-catalyzed conditions discovered for indole, we found that aniline was a competent nucleophile. Specifically, aziridine 3 undergoes preferential attack by the nitrogen atom of 12 to produce diamine derivative 13 in high yield with nearly complete transfer of stereochemical information (Scheme 4). The differentially substituted, chiral 1,2-diamine motif will be valuable for bio-active small molecule synthesis.
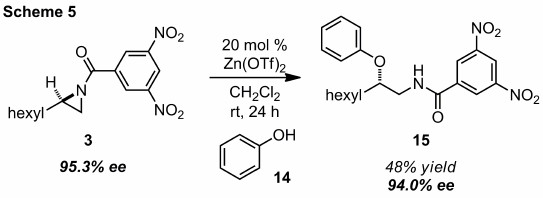
The consistent preference for stereospecific ring opening displayed by the N-acylaziridines incited further evaluation of synthetically useful heteroatomic nucleophiles. We have briefly investigated phenols as interesting substrates since a large number of commercially available pharmaceuticals contain this functionality. Our success in the kinetic resolution with BINOL operating as the nucleophile strongly suggested phenols would participate in ring opening. Indeed, zinc triflate will also catalyze the enantiospecific addition of phenol (14) to N-acylaziridine 3 in moderate yield without loss of enantiopurity to produce amino ether 15 (Scheme 5).
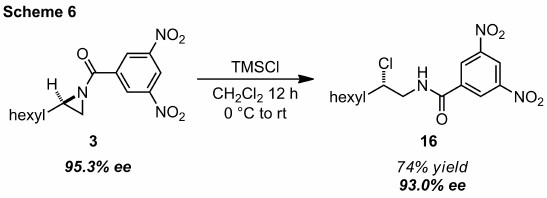
Synthesis of stereodefined halogen–carbon bonds has been a recent area of interest in organic chemistry. Treatment of aziridine 3 with excess trimethylsilyl chloride (TMSCl) generated 1,2-chloroamide 16 in good yield (Scheme 6). Consistent with previous nucleophiles, addition occurred stereospecifically with minimal loss of ee. These data collectively support the general concept that terminal DNB-protected aziridines possess inherent preference for nucleophilic attack at the more hindered carbon. This precedent is intriguing as other TMS reagents are commercially available with not only halide nucleophiles, but also nitrogen (azide) and carbon (cyanide) nucleophiles as well.
In conclusion, support from the Petroleum Research Fund has facilitated the development of N-acylaziridines as linchpin intermediates for the synthesis of nitrogen-containing small molecules. The preference for attack at the more hindered aziridine carbon in an enantiospecific fashion is important for application of these methods in organic synthesis. During the current grant period, three (3) Master's students and (4) undergraduate students gained valuable research experience on this project. We will continue to develop novel reactions based on this work though continued PRF support, and this data is being used in applications for expanded research funding through the National Institutes of Health and National Science Foundation.









