Reports: DNI350840-DNI3: Bio-inspired Inorganic Catalysts for CO2 Reconversion
 printer friendly
printer friendlyReduction of CO2 is of great interest because the reduced forms of CO2 such as formic acid, methanol, or methane can be directly used as fuels, which establishes CO2 as an abundant and inexpensive source for alternative energy. Although there have been continuing efforts in developing coordination complexes that can catalyze the reduction of CO2,1 none has shown the comparable efficiency and selectivity of the CO2-reducing enzymes found in nature.
Formate dehydrogenase (FDHs) are enzymes that catalyze the two electron oxidation of formate (HCO2–) to CO2 coupled to the reduction of NAD(P)+ to NAD(P)H.2 In some prokaryotes, the FDHs contain molybdenum (Mo) or tungsten (W) cofactors, by which an efficient CO2-reductase activity has been also observed. Recently, Hirst and coworkers have shown that a purified W-FDH can activate an electrode to reduce CO2 to formate more efficiently than any of known synthetic catalysts.3
The X-ray structures of several Mo or W containing FDHs are known,4 in which the metal ion is in a square pyramid geometry with two equatorial pyranopterins and an –SH (or –OH) axial ligand in the reduced state, Figure 1. In light of the known FDH structures as well as the efficient electrocatalytic activity of W-FDH, we have studied on the CO2 reactivity of a synthetic model complex of W-FDH.5
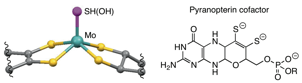
Figure 1. Active site structures in the reduced Mo-FDH (E. coli)4a.
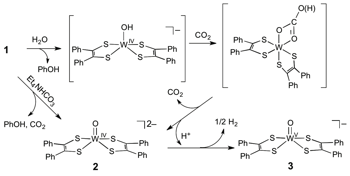
Holm has previously reported6 that a group of phenolate bound tungsten or molybdenum bis(dithiolene) complexes such as [WIV(OPh)(S2C2Me2)2]– undergo hydrolysis in the presence of excess water to yield WIV=O complexes via formation of {WIV-OH} species. These reports inspired us to explore CO2 reactivity of [WIV(OH)(S2C2Ph2)2]– which can be prepared in situ through hydrolysis of [WIV(OPh)(S2C2Ph2)2]– (1) in the presence of water.
We first sought a hydrolysis condition that can be achieved with the minimum amount of water possible. We found that a relatively small amount of water was sufficient enough to induce hydrolysis of [W(OPh)(S2C2Ph2)2]– (1) to yield [WIV(O)(S2C2Ph2)2]2– (2). However, we noticed that 2 was not stable and it slowly converts into another well known compound, [WV(O)(S2C2Ph2)2]– (3) (Scheme 1), which is associated with the reduction of H+ to hydrogen.
Scheme 1

IR spectroscopy supports the generation of 3 as the final hydrolysis product as well as the released free phenol, Figure 2. The 18OH2 isotope experiment indicates that the oxygen atoms in the reaction products originate from water through hydrolysis of [WIV(OPh)(S2C2Ph2)2]– (1), during which the intermediate [WIV(OH)(S2C2Ph2)2]– must have been generated. The observed 937 cm–1 peak from the crude reaction products is assigned to the stretching frequency of WV=O in 3, while the additional peak at 915 cm–1 is due to the hydrogen bonded WV=O moiety to free phenol that is released from 1.
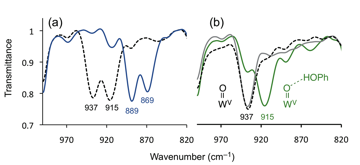
Figure 2. IR spectra (Nujol) of (a) the crude reaction products of [WIV(OPh)(S2C2Ph2)2] (1) and 10 equiv. of H2O (black dotted) and those of 1 and 10 equiv. of H218O (blue solid), (b) the isolated reaction product [WV(O)(S2C2Ph2)2]– (3) (black dotted), 3 in the presence of 2 equiv of externally added phenol (green solid), and re-isolated 3 after removing phenol by precipitation (grey solid).
Next, we studied the CO2 reactivity of [WIV(OPh)(S2C2Ph2)2]– (1) in the presence of water. The reaction of 1/CO2/H2O does not exhibit the anticipated FDH activity. Instead, [WIV(OH)(S2C2Ph2)2]– reacts with CO2 to form a presumable (bi)carbonato intermediate which immediately undergoes decarboxylation to generate [WIV(O)(S2C2Ph2)2]– (2) and CO2 (Scheme 2). The formation of (bi)carbonato species from CO2 with a metal-hydroxide is common for many transition metal complexes. In order to evaluate the prospect of the proposed (bi)carbonato intermediate during the reaction (Scheme 2), [WIV(OPh)(S2C2Ph2)2]– (1) was reacted with one equiv. of Et4NHCO3. Upon addition of bicarbonate to 1, the oxo compound 2 formed immediately, as monitored by UV-Vis spectroscopy. The result supports a notion that the O atom of CO2 is inserted into 2 through a (bi)carbonate-bound W intermediate.
Scheme 2
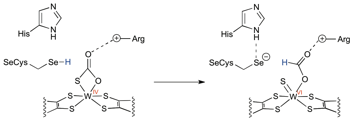
The FDHs utilize a bis(dithiolene) bound MIV (where M = Mo or W) ion to reduce CO2 to formate. The use of higher-valent metal ions with non-innocent ligands by the enzyme is a very different strategy from most synthetic systems in which the metal ions in much lower valences, 0, +1, and +2, are utilized to activate CO2. Our current study shows that the presence of a nucleophilic ligand such as OH– is crucial to initiate CO2 reactivity with a bis(dithiolene) tungsten(IV) species to likely form a tungsten (bi)carbonate intermediate. Indeed, a recent calculation study on the mechanism of Mo-FDH suggests that a MoIV-thiocarbonato intermediate forms during the oxidation cycle of formate to CO2. With respect to the reverse reaction by W-FDH, one can expect the conversion of WIV-thiocarbonate species to WVI-formate (Scheme 3), similar to the typical O-atom abstraction chemistry known for this class of enzyme. In our current synthetic model system, however, we were not able to imitate the reduction step. This may be due to the lack of a proton delivery channel near the metal active site, i.e., selenocysteine and histidine, that is strictly conserved in the FDH proteins. Future studies will focus on the factors critical to induce an O-atom abstraction from a putative tungsten-(bi)carbonate intermediate over the oxide (O2–) abstraction chemistry observed.
Scheme 3
References:
(1) Sakakura, T.; Choi, J. C.; Yasuda, H. Chem. Rev. 2007, 107, 2365-2387.
(2) Hille, R. Chem. Rev. 1996, 96, 2757-2816.
(3) Reda, T.; Plugge, C. M.; Abram, N. J.; Hirst, J. Proc. Natl. Acad. Sci. U. S. A. 2008, 105, 10654-10658.
(4) (a) Raaijmakers, H. C.; Romao, M. J. J. Biol. Inorg. Chem. 2006, 11, 849-854. (b) Raaijmakers, H.; Macieira, S.; Dias, J. M.; Teixeira, S.; Bursakov, S.; Huber, R.; Moura, J. J.; Moura, I.; Romao, M. J. Structure 2002, 10, 1261-1272.
(5) Seo, J.; Kim, E. Inorg. Chem. 2012, 51, 7951-7953.
(6) Sung, K. M.; Holm, R. H. J. Am. Chem. Soc. 2001, 123, 1931-1943.









