Reports: ND649930-ND6: Energetics of the Ligand and Solvent Coordination of Catalytically Important Organometallic Complexes
 printer friendly
printer friendlyIntroduction
Using the recently constructed imaging Photoelectron Photoion Coincidence (iPEPICO) experiment of the VUV beamline of the SLS synchrotron, we have continued working on projects to determine highly accurate bond energies in the gas phase, with special emphasis on metal-ligand bonds. In the second funding cycle, we have collected more experimental data on a number of small and medium-size systems and have published a number of papers on the PEPICO experiments of smaller molecules as well as organometallics. Very recently, we have also built and published a new, double-imaging PEPICO (i2PEPICO) experiment to study dissociative photoionization dynamics.
Small molecule iPEPICO studies
The unimolecular dissociation of internal energy selected C3H5Br+ isomer ions was studied by iPEPICO yielding accurate dissociation onsets to Br-loss reactions in the molecular ion. Quantum chemistry was used to map out the potential energy surface connecting the isomer cations, and to determine the dissociation mechanism of Br loss to the two C3H5+ isomer fragment ions. The results showed that cis- and trans-1-bromopropenes, 3-bromopropene and bromocyclopropane dissociate into the allyl cation, whereas 2-bromopropene dissociates into the 2-propenyl cation. There of the systems were found to be metastable. This slow dissociation is due to the deep potential energy well, leading to a larger density of states at the threshold. The dissociation kinetics of 1-bromopropene cations starts with isomerization to allyl bromide cation prior to dissociation.
The H/D loss and CH3/CD3 loss reactions from four energy selected ethanol isotopologue ions have been measured by iPEPICO spectroscopy. In the lowest energy dissociation channel, the α-carbon loses a hydrogen or deuterium atom. Asymmetry in the daughter ion time-of-flight peaks, ab initio study of the reaction rates, and shifts in the phenomenological onsets between isotopologues revealed that H/D loss is slow at its onset. Tunneling through a reverse barrier along the reaction coordinate was found to play a significant role. The higher energy methyl loss channel appears at its thermochemical threshold and was successfully modeled for all 4 isotopologues using the simplified statistical adiabatic channel model up to the crossover. However, modeling the higher energy H-loss to methyl-loss ratio proved to be more challenging as the H-loss signal never disappears completely, and the persistent H-loss signal can either be due to a non-statistical process or a statistical one that cannot be described by any of the statistical rate theories tested herein. The shape of the photoelectron spectrum as well as our ab initio calculations indicate that the lowest energy ethanol ion structure lies considerably below the literature ionization energy.

Organometallics
As a follow-up on a recent PEPICO study (outlined in last year's report), in collaboration with the Armentrout group, seven metallocene ions (Cp2M+, M = Ti, V, Cr, Mn, Fe, Co, Ni) were studied by threshold collision induced dissociation (TCID) guided ion-beam mass spectrometry to derive accurate ionic metal-cyclopentadienyl bond dissociation energies. For all seven metallocene ions, the dominant dissociation pathway is simple Cp loss from the metal while traces of other fragment ions were also detected, such as C10H10+, C10H8+, C8H8+, C3H3+, H2M+, C3H3M+, C6H6M+, and C7H6M+, depending on the metal center. Statistical modeling of the Cp-loss TCID experimental data allowed the extraction of 0 K [CpM+– Cp] BDEs.
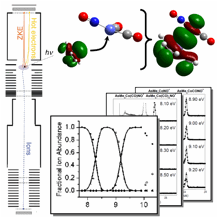
In last year's report, we had outlined a study on phosphine and phosphine-analog cobalt-organic complexes. Recently, we have finished the analysis of the data and published this project in Organometallics. With the help of DFT and statistical rate calculations, the first two, metal-carbonyl bond dissociation energies of the gas-phase organometallic complex ions were determined. These values, along with ionization energies from photoelectron spectra were used to discuss the electron acceptor and electron donor properties of the two phosphine and two phosphine-analog ligands and it was shown that, using the experimental bond energies and the first ionization energies, both the overall electron-donor properties of the ligands as well as its decomposition to σ and ¹-type interactions can be assessed. Our numbers yield experimental evidence that, with regards to the bond strength, the ¹-acidity of the ER3 ligands and, consequently, the ¹-back-donation interaction between the transition metal and the carbonyl ligand is of primary importance.
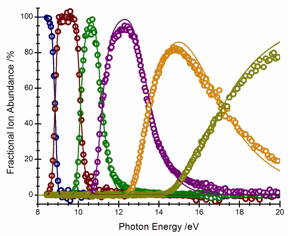
The dissociation dynamics of energy-selected iron carbonyl cations have been investigated using iPEPICO. The dissociation of the molecular ion proceeds by five sequential carbonyl-loss steps in the energy range from 8.5 – 20 eV. The experimental breakdown diagram and asymmetric time-of-flight (TOF) distributions were modeled using RRKM theory for the dissociation rates while the energy distributions were modeled using statistical microcanonical product energy distributions. For the first three carbonyl-loss dissociations, the calculated energy distribution show near-perfect agreement with the experimental breakdown curves, modeling well the statistical broadening of the energy distribution functions. Beyond a photon energy of 12 eV, the calculated shape of the breakdown curves start diverging from the experimental data, possibly indicating that the high-energy parts of the distributions exhibit non-statistical dynamics. Chromium hexacarbonyl was also investigated, where we measured six consecutive carbonyl-loss dissociations from the molecular ion. The modeling of this data is currently underway.
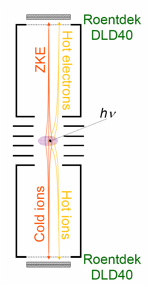
Instrument Development
During the 2012 spring PRF-funded trip to the Swiss Light Source, we have built, in addition to the original iPEPICO endstation, a second, complementary double imaging i2PEPICO experiment. Volatile samples can be introduced at room temperature or in a molecular beam, a pyrolysis source allows for radical production, and non-volatile solids can be evaporated in a heated cell. Monochromatic VUV radiation ionizes the sample and both photoelectrons and photoions are velocity map imaged onto two fast position sensitive detectors and detected in delayed coincidence. For this new setup, we have developed new data acquisition and processing approaches to record coincidence processes at very high rates. The setup will be used to extract kinetic energy release distributions upon dissociative photoionization and is capable of resolving pulsed molecular beam profiles to separate the signal from the beam-cooled species from the background effusive signal.