Reports: DNI549596-DNI5: Structure and Dynamics of Aqueous and Aqueous-Hydrocarbon Fluids between Charged Surfaces
 printer friendly
printer friendlyThe project was continued in the first no-cost-extension year to support a PhD student to study the clay swelling in water-methane mixture under different temperature/pressure sedimentary conditions. A postdoctoral researcher also participated part of research. The PI studied the hydration force mechanism when mica clays was immersed in KCl electrolyte, and published one paper in Langmuir. Below is a summery of students work that impacted their education and training in molecular simulations.
Clay swelling at different sedimentary basin conditions was studied in previous works1-3. The present project focuses on the clay swelling mechanism in aqueous-hydrocarbon mixture and the adsorption structure of water, ion and hydrocarbon species near charged clay surfaces. The potential model used for water molecules is the rigid SPC model4. The chemical potential of pure water at 298K is found to be -45.56 kJ/mol. The model of clay used in this work is the sodium-saturated Wyoming-type montmorillonite that has the chemical formula Na0.75[Si7.75Al0.25](Al3.5Mg0.5)O20(OH)4. The CLAYFF6 force field was used in this work. The swelling curve obtained from the GCMC simulations is in close agreement with experimental results7, as well as with early molecular dynamics (MD) simulation results6. These results are shown in Fig. 1.
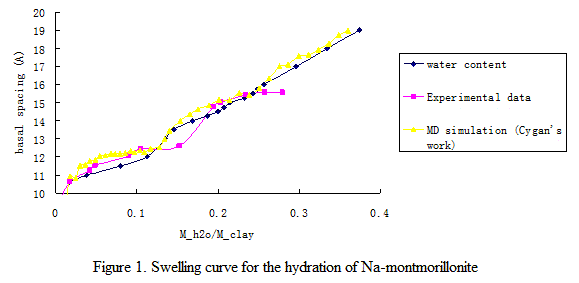
Figure 1. Swelling curve for the hydration of Na-montmorillonite
Previous work8-10 studied the structure and dynamics of water-methane system. However, it is not clear how many water and methane molecules should be set under specific P/T conditions. In this work, a series of NPT Monte Carlo simulations were performed to compute the chemical potentials of water and methane in the mixture in gas phase under different T/Ps. The pressure of water is set to the value of saturated pressure corresponding to the mixture temperature. The pressure of methane is set to the value equal to the rest of the total pressure11. Molecular model for methane is represented by the rigid OPLS-AA12 model. Widom's insertion method13 is used to compute chemical potential of a fluid in the NPT ensemble. This method includes insertions of a test particle into the simulation box equilibrated at the desired temperature and pressure with the Monte Carlo simulation in the NPT ensemble. The energy of interaction is used to calculate the chemical potential of the fluid, i.e.14
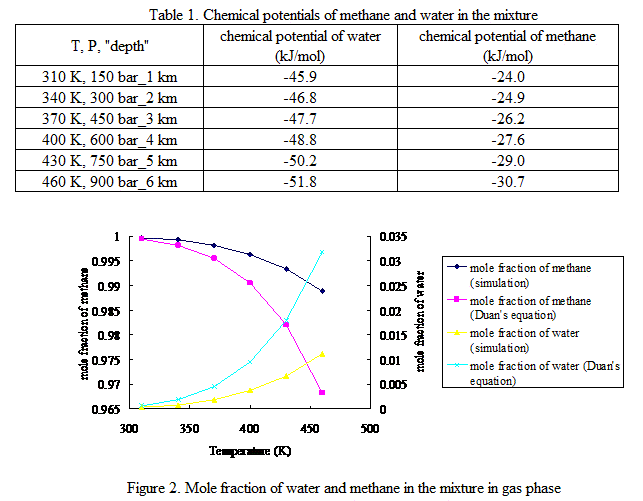
The NPT Monte Carlo simulation results of chemical potentials of water and methane under different P/T conditions are listed in Table 1. To validate this method, we calculated the mole fraction of water and methane in the mixture in gas phase under those P/T conditions with corresponding chemical potentials of water and methane via GCMC simulation. The results are compared with results from a semi-empirical equation15 in Fig. 2.
Table 1. Chemical potentials of methane and water in the mixture
T, P, "depth" | chemical potential of water (kJ/mol) | chemical potential of methane (kJ/mol) |
310 K, 150 bar_1 km | -45.9 | -24.0 |
340 K, 300 bar_2 km | -46.8 | -24.9 |
370 K, 450 bar_3 km | -47.7 | -26.2 |
400 K, 600 bar_4 km | -48.8 | -27.6 |
430 K, 750 bar_5 km | -50.2 | -29.0 |
460 K, 900 bar_6 km | -51.8 | -30.7 |
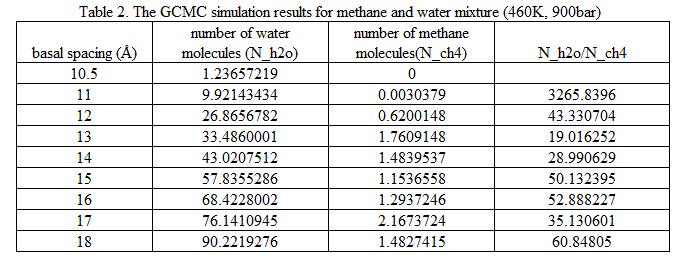
Figure 2. Mole fraction of water and methane in the mixture in gas phase
Using the GCMC simulation method, we have studied the clay swelling in water-methane mixture at 460 K and 900 bar, corresponding to the temperature and pressure at burial depth of 6 km. The chemical potentials of water and methane evaluated in NPT Monte Carlo simulations are used in those GCMC simulations. A series of GCMC simulations were performed with various basal spacing from 10.5 � to 18 �. The average numbers of water molecules and methane molecules entering the clay interlayer were calculated and listed in Table 2. Under this P/T condition, water molecules enter clay interlayer first. Methane molecules cannot enter clay interlayer until the basal spacing reach 12 �. The molecular number ratio of water to methane varies significantly versus the basal spacing, demonstrating that clay surface gap has remarkable influence on the formation of methane hydrate. Future molecular dynamics simulations based on the GCMC simulation results are needed to investigate the stability of those methane hydrate structures.
Table 2. The GCMC simulation results for methane and water mixture (460K, 900bar)
basal spacing (�) | number of water molecules (N_h2o) | number of methane molecules(N_ch4) | N_h2o/N_ch4 |
10.5 | 1.23657219 | 0 |
|
11 | 9.92143434 | 0.0030379 | 3265.8396 |
12 | 26.8656782 | 0.6200148 | 43.330704 |
13 | 33.4860001 | 1.7609148 | 19.016252 |
14 | 43.0207512 | 1.4839537 | 28.990629 |
15 | 57.8355286 | 1.1536558 | 50.132395 |
16 | 68.4228002 | 1.2937246 | 52.888227 |
17 | 76.1410945 | 2.1673724 | 35.130601 |
18 | 90.2219276 | 1.4827415 | 60.84805 |
(1) Sposito, G.; Skipper, N. T.; Sutton, R.; Park, S. H.; Soper, A. K.; Greathouse, J. A. Proceedings of the National Academy of Sciences of the United States of America 1999, 96, 3358-3364.
(2) Skipper, N. T.; Lock, P. A.; Titiloye, J. O.; Swenson, J.; Mirza, Z. A.; Howells, W. S.; Fernandez-Alonso, F. Chemical Geology 2006, 230, 182-196.
(3) Cygan, R.T.; Guggenheim, S.; van Groos, A. F. K. Journal of Physical Chemistry B 2004, 108, 15141-15149.
(4) Teleman, O.; Jonsson, B.; Engstrom, S. Molecular Physics 1987, 60, 193-203.
(5) Tambach, T. J.; Bolhuis, P.G.; Hensen, E. J. M.; Smit, B. Langmuir 2006, 22, 1223-1234.
(6) Cygan, R.T.; Liang, J. J.; Kalinichev, A. G. Journal of Physical Chemistry B 2004, 108, 1255-1266.
(7) Fu, M. H.; Zhang, Z. Z.; Low, P. F. Clays and Clay minerals 1990, 38, 485-492.
(8) Park, S. H.; Sposito, G. Journal of Physical Chemistry B 2003, 107, 2281-2290.
(9) Titiloye, J. O.; Skipper, N. T. Molecular Physics 2001, 99, 899-906.
(10) Titiloye, J. O.; Skipper, N. T. Journal of Colloid and Interface Science 2005, 282, 422-427.
(11) Duan, Z., Moller, N., Weare, J.H. Geochimica et Cosmochimica Acta 1992, 56, 1451-1460.
(12) Jorgensen, W. L., Maxwell, D. S., Tirado-Rives, J. Journal of the American Chemical Society 1996, 118, 11225-11236.
(13) Allen, M. P., Tildesley, D. J. Computer Simulation of Liquids Oxford University Press, 1987.
(14) Frenkel, D., Smit, B. Understanding Molecular Simulation Academic, New York, 1996.
(15) Duan, Z., Mao S. Geochimica et Cosmochimica Acta 2006, 70, 3369-3386.









