Reports: UR151369-UR1: Utilizing Halogenated Carbonyls to Direct Stereoselective Synthesis
 printer friendly
printer friendlyACS-PFR-UR Narrative Progress Report
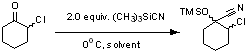
During our first year of support from ACSPRF–UR an investigation into thestereoselective addition of trimethylsilyl cyanide (TMSCN) to a variety of halogenated (chloro- & -fluoro) carbonyls (ketones & imines). These reactions work extremely well and in select cases are completely selective.
TMSCN Additions to α-Chloroketones:
Our laboratory has been investigating the TMSCN addition to α-fluoroketones in high yield and diastereoselectivity. This past year has focused on optimizing reaction conditions and substrate scope. The reaction has been optimized by investigating both the solvent and reaction temperature (Table 1). A variety of solvents were investigated using 2-chlorocyclohexanone as
Table 1: Effect of Solvent on %Conversion and Selectivity in TMSCN Mediated Additions to 2-Chlorocyclohexanone
Entry | Solvent | % Conversiona | dr |
1 | CH2Cl2 | 75 | 1:1 |
2 | THF | 98 | 2:1 |
3 | EtOAc | 66 | 2:1 |
4 | Toluene | 50 | 2:1 |
5 | CH3CN | 100 | 2:1 |
6 | DMF | 100 | 5:1 |
7 | DMF/CH3CN (3:1) | 100 | 3:1 |
8 | DMF/CH3CN (1:3) | 100 | 2:1 |
9 | DMF/CH3CN (1:1) | 100 | 6:1 |
a% Conversion and dr were determined using GC and/or NMR.
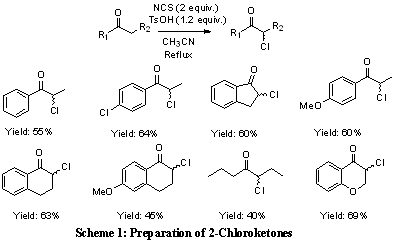
the test substrate. The reaction only went to completion using dimethylformamide (DMF) and acetonitirile (CH3CN) as the solvent. The diastereoselectivity increased from 2:1 to 5:1 using DMF in comparison to CH3CN. The optimal reaction conditions were determined to be a mixture of DMF:CH3CN (1:1) which resulted in complete conversion and dr of 6:1. To further examine the scope and limitations of this methodology a variety of 2-chloroketones were prepared using previously established methods. The preparation of the 2-chloroketones was accomplished by refluxing the ketone with N-chlorosuccinimide under acidic conditions in CH3CN (Scheme 1). Using this method our laboratory was been able to prepare a variety of 2-chloroketones in good yield (40-69%).
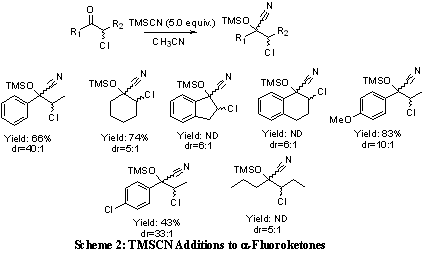
With the 2-chloroketones in hand the substrate scope of the TMSCN additions was investigated. It was discovered that the reaction worked well for a variety of substrates with isolated yields as high as 83% and selectivities up to 40:1 (Scheme 2). These reactions usually proceed in 100% conversion however product is lost to degradation during purification.
Our focus is currently on determining the absolute stereochemistry by starting with chiral α-chloroketone. These can be prepared using methodology developed by Jorgenson and co-workers. With the chiral starting material in hand the TMSCN addition will be initiated. The product will then be used to determine the absolute stereochemistry of the major and minor stereoisomers.
The second area of focus has been the TMSCN addition to α-fluorinated ketones. This past year has focused on investigating substrate scope of the TMSCN addition to fluorinated ketones. These reactions proceed on good yield (up to 95%) and modest to good selectivity (3:1 to 22:1 cis/trans) (Scheme 3). This method was tolerant for a variety of functional groups as well as electron withdrawing and donating groups.
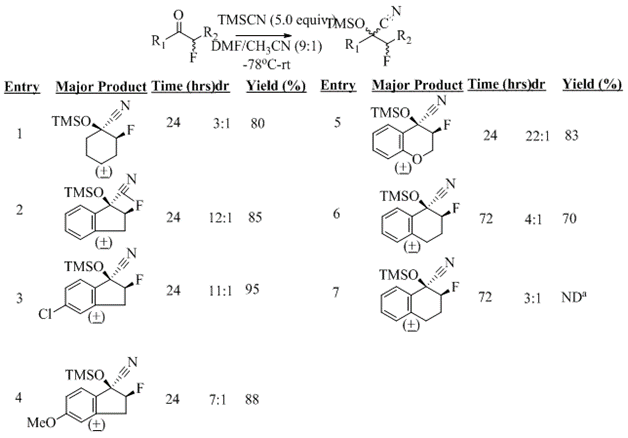
Scheme 3: TMSCN Additions to α-Fluorinated Ketones
The stereochemistries of the cyclic products were determined by examining their 13C and 19F NMR spectra. It has been established that the coupling constants of nitrile carbons with fluorine (3JC-F) are quite different in the cis (predicted Jz =8.0Hz) versus trans (predicted Jz =1.8Hz)orientations (Figure 1).9 Based on our observed coupling constant, the major product for the cyclohexanone derivative (Table 1, entry 1) is the cis- isomer (3JC-F= 5.1 Hz) and the minor isomer is the trans- stereochemistry (3JC-F= 2.1Hz) (Figure 1) in accordance with previously published reports. The 19F NMR spectra also show a significant chemical shift difference between the cis- versus trans- stereoisomers. In the case of the of the cis- diastereomer of the cyclohexanone derivatives, a 19F NMR absorption appears at -185.7. ppm, while the trans- absorbs at -179.8 ppm.
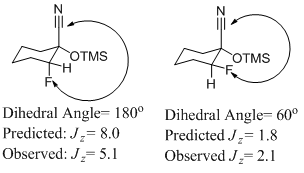
Figure 1: Coupling constants for the cis- and trans stereoisomer's of the derivatized cyclohexanone product
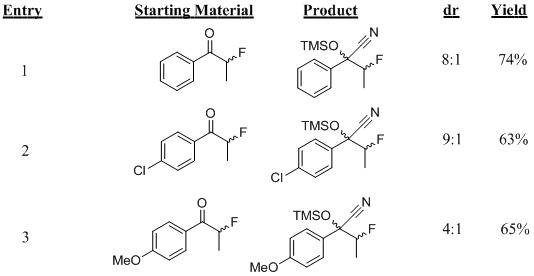
The TMSCN addition also worked well for acyclic α-fluorinated ketones, proceeding in yields as high as 74% (Table 3). The reaction proceeded with both the electron withdrawing and donating substituents in good yields (Table 3, entries 2 & 3). Due to the difficulties associated with the preparation of chiral acyclic α-fluorinated ketones, the absolute stereochemistry of the products has yet to be determined and is currently under investigation.
Table 3: TMSCN additions to acyclic α-fluoroketones
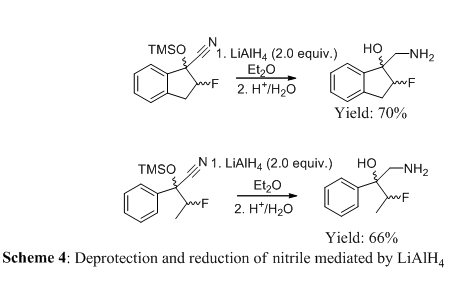
In order to perform purification via chromatographic methods, the cyano- products were reduced with LiAlH4 and deprotected to form the corresponding 1,2-amino alcohols in good yield (Scheme 4). It is important to note that the silylenol ether bond of the initial product was extremely labile, leading to a retro reaction and the formation of the starting material upon purification by chromatography (acidic, neutral, and basic conditions). These products were stable to chromatography and were purified using both flash chromatography. 1,2-amino alcohols have been key structural components for a variety of drug candidates and natural product targets. This newly developed methodology serves as a useful alternative to traditional preparations such as the ring opening of epoxides or aziridines.
Currently, we are in the process of performing X-ray crystallography to determine the relative stereochemistry of the acyclic products and these results will be reported in due course.
Summary: Our laboratory has begun to open a new library of stereoselective additions to halogenated ketones in good yield and selectivity. These newly developed methods will serve as important precursors for a variety of biologically active molecules. Our next step will to develop a firm mechanistic understanding of the reaction in order to increase yield and improve selectivity in these reactions.
References:
1.) Lee, J. C.; Bae, Y.; Chang, S. Efficient a-Halogenation of Carbonyl Compounds by N-Bromosuccinimide and N-Chlorosuccinimde Bulletin of the Korean Chemical Society 2003, 24,
2.) Marigo, M.; Bachmann,S.; Halland, N.; Braunton, A.; Jørgensen, K. A. Highly Enantioselective Direct Organocatalytic α-Chlorination of KetonesAngewandte Chemie International Edition, 2004, 43, 5507-5510.









