Reports: DNI350287-DNI3: Physical Properties and Reactivities of Peroxomanganese (III) Complexes
 printer friendly
printer friendlyThe subject of this award is to contribute to an in-depth understanding of the initial steps in hydrogen peroxide activation for manganese catalysts by defining the physical and reactivity properties of peroxomanganese(III) intermediates, which are proposed to be among the first species formed when manganese complexes react with hydrogen peroxide. Such catalytic systems are of interest because manganese centers and hydrogen peroxide mediate important oxidative transformations, including those involved in the functionalization of petroleum feedstocks and fabric and paper bleaching. In year two of this project we have i) published a detailed study comparing geometric and electronic stuctures of peroxomanganese(III) complexes supported by tetradentate aminopyridyl and aminoquinolinyl ligands, ii) designed and synthesized Mn(II) complexes supported by new aminoquinoline ligands expected to be more oxidatively robust than their first-generation analogues, and iii) elucidated the reaction landscape of a manganese(II) complex with superoxide and hydrogen peroxide, which features the conversion of a peroxomanganese(III) adduct to a bis(m-oxo)dimanganese(III,IV) species.
In total, this two-year award has supported the training of three graduate students and has resulted in three publications to date, positively impacting my career as an assistant professor.
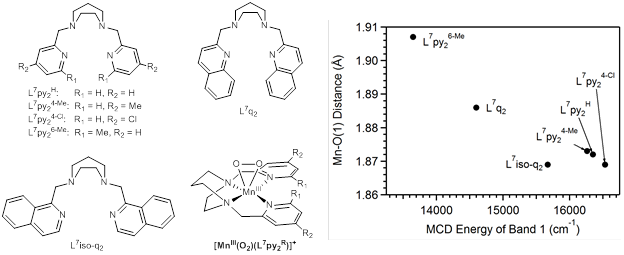
Figure 1. Ligands used to support peroxomanganese(III) complexes, representative structure of a [MnIII(O2)(L7py2R)]+ complex, and correlation of Mn-O(peroxo) distance with lowest-energy MCD transition.
In this year, we completed our comparison of MnIII-O2 adducts supported by tetradentate ligands with systematically varied steric and electronic properties (Geiger, R. A. et al. Eur. J. Inorg. Chem. 2012, 1598-1608.). Magnetic circular dichroism (MCD) and density functional theory (DFT) studies of these six complexes establish a correlation between the computed Mn-O(peroxo) bond length and the position of the lowest-energy MCD band (band 1; see Figure 1). Notably, this correlation is attributed to the steric properties of the supporting ligand; electronic perturbations among this series have a limited effect. For example, pyridine substituents (R = Cl, H, and Me) in the 4-position hardly affect the energy of band 1, whereas steric interactions between pyridine or quinoline groups (e.g., for the L7py26-Me and L7q2 ligands) lead to a red-shift of band 1 that is attributed to an elongation of one Mn-O(peroxo) bond (Figure 1, far right). We intend to exploit this tuning of the Mn-O(peroxo) interaction to activate MnIII-O2 species using sterically encumbered ligands. These results also suggest that enzyme active sites may prevent formation of symmetric MnIII-O2 adducts (i.e., with near equivalent Mn-O(peroxo) distances) by putting strict steric constraints on the peroxo binding pocket. Presumably, asymmetric MnIII-O2 spcies with an elongated Mn-O(peroxo) bond would be more reactive towards electrophilic substrates, as previously suggested (Annaraj, J. et al. Angew. Chem. Int. Ed. 2009, 48, 4150-4153).
Our previous work has established that the peroxomanganese(III) adducts supported by our L7py2R family of ligands are quite electrophilic (Geiger, R. A. et al. Dalton Trans. 2011, 40, 1707-1715.). However, we have been unable to trap any intermediates when these compounds are treated with acids or acyl chlorides. Instead we observe the immediate disappearance of the peroxomanganese(III) chromophore and the formation of manganese(II) species. Acids or acyl chloride reagents are expected to react with the MnIII-O2 unit to generate MnIII-OOH or MnIII-OOR species, respectively, which could subsequently undergo O-O cleavage to yield high-valent oxomanganese species. Although such reactions have been observed for corresponding iron(III) complexes, such chemistry has remained quite elusive for manganese(III) centers. For our complexes, we propose that the peroxomanganese(III) species reacts with acids or acyl chlorides to yield a strong oxidant that initiates decomposition of the L7py2R ligand. In support, Groni et al. have observed that the peroxomanganese(III) complex [MnIII(O2)(mL52)]+ (mL52 = N-methyl-N,N¢,N¢-tris(2-pyridylmethyl)ethane-1,2-diamine) decays upon treatment with acid to yield a Mn(II) center bound to two pyridinecarbyoxylate ligands that are formed by oxidation of mL52 (Groni, S. et al. Inorg. Chem. 2008, 47, 3166-3172). Thus, we reasonably suspect that the methyl linkers of the L7py2R ligands could be susceptible to oxidization by high-valent oxomanganese species. Accordingly, we have synthesized the L7BQ and L8BQ ligands, where the quinoline rings are attached directly to the 1,4-diazepane and 1,5-diazacyclooctane fragments, respectively (Figure 2). The crystal structure of the [MnII(L7BQ)(OTf)2] complex (Figure 2) shows the tetradentate ligand bound in the trans fashion, similar to that of the L7py2R ligands. We are currently in the process of characterizing the peroxomanganese(III) complexes supported by the BQL7 and BQL8 ligands and exploring their activation by acids and acyl chlorides. We note that the putative [MnIII(O2)(L7BQ)]+ complex shows enhanced thermal stability relative to [MnIII(O2)(L7q2)]+.
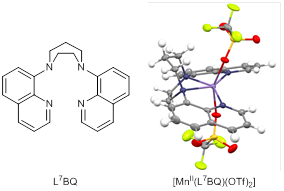
Figure 2. Structures of the L7BQ ligand (left) and an ORTEP diagram of the [MnII(L7BQ)(OTf)2] complex (right).
In Year 1 of this project, we described the geometric and electronic structures of a peroxomanganese(III) unit supported by the pentadentate N4py ligand (N4py = N,N-bis(2-pyridylmethyl)-N-bis(2-pyridyl)methylamine). This complex was generated by treatment of [MnII(N4py)(OTf)2] with KO2 in acetonitrile (Scheme 1). Unlike all other peroxomanganese(III) complexes reported to date, [MnIII(O2)(N4py)]+ can only be generated using KO2. If [MnII(N4py)(OTf)2] is treated with H2O2 and Et3N in acetonitrile, the dinuclear [MnIIIMnIV(m-O)2(N4py)2]3+ complex is formed even at low temperatures. When [MnIII(O2)(N4py)]+ is formed from [MnII(N4py)(OTf)2] and KO2 in high yields, it decays to a mixture of manganese(II) species. However, if it is formed in only 50% yield or less, it converts to the [MnIIIMnIV(m-O)2(N4py)2]3+ complex. Separate experiments have shown that treatment of [MnIII(O2)(N4py)]+ with [MnII(N4py)(OTf)] also leads to the formation of [MnIIIMnIV(m-O)2(N4py)2]3+. Thus, the MnII-N4py system shows a complex reaction landscape, featuring a variety of intermediates. Because dissociation of a pyridylmethyl arm is require for formation of [MnIII(O2)(N4py)]+, our current hypothesis is that the rate of pyridine dissociation influences whether or not the MnII center reacts with an oxidant to afford a MnIII-O2 or MnIIIMnIV dimer. Understanding the complex factors that govern the relative rates of formation of these intermediates is important given that oxo-bridged MnIIIMnIV dimers are often viewed as "dead-end" intermediates due to their high thermodynamic stability. Understanding how to disfavor the formation of such species, could be an important step in accessing more reactive intermediates.
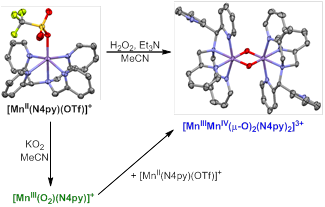
Scheme 1. Experimentally observed reaction landscape for [MnII(N4py)(OTf)]+ in the presence of H2O2 or KO2.









