Reports: ND150808-ND1: New Studies in Alkene 'Pseudohalogen' Difunctionalization
 printer friendly
printer friendlyNarrative Report Text. The enantioselective triazolination chemistry his was a central aim of our proposal and was the most well developed in terms of preliminary results. We spent a significant amount of time optimizing a Pt(II) catalyzed triazolination of monosubstituted alkenes using several chiral ligands; after a substantial period of investigation, the best conditions discovered to date include a slow addition (24 h) of iodane 2 to styrene and 5 mol% (R)-or (S)-DIFLUORPHOS¥PtCl2 catalyst 3 in refluxing THF. To our gratification, the desired triazoline product 3 (as a mixture of interconverting tautomers) was formed in 65% yield and in 90% ee (Scheme 1). We assembled a table of substrates that work well in 39-76% yield and 84-92% ee.

Scheme 1.
However, a serious problem arose. On several occasions, the iodane reagent 2 exploded when it came into contact with the Pt metal catalyst in solution. Under the circumstances, we found it difficult to proceed with the chemistry using the iodane 2. We then sought alternative sources of electrophilic azide that would be safer and more reliable. We achieved a modest degree of success by employing TMSN3 as the azide source, CuI as the metal catalyst, and Selectfluor as an oxidant. When we employed 1-heptene as a substrate, we quickly realized that a reaction byproduct 6was potentially as if not more interesting than the desired process (Scheme 2).

Scheme 2.
The selective incorporation of fluorine into organic molecules has advanced in dramatic ways over the last 30 years. Although arene and alkyne fluorination using metalcatalysis has received much attention, corresponding methods for metal-catalyzed alkane fluorination remain only a promising goal (Scheme 3). To date, the most notable methods for alkane fluorination involve the use of stoichiometric quantities of difficult-to-handle or indiscriminate reagents such as elemental fluorine, cobalt trifluoride (polyfluorination), or potentially explosive cesium fluoroxysulfate. The result in Scheme 2 interested us in the question of whether alkane fluorination could indeed be selectively catalyzed under mild conditions. What is more, a mild procedure using safe, commercially available alternatives would be complementary to pioneering methods, and perhaps be applicable to closely related allylic and benzylic substrates. We used these preliminary results to develop the fluorination of a series of aliphatic, benzylic, and allylic substrates by a polycomponent catalytic system involving commercially available Selectfluor, putative radical precursor N-hydroxyphthalimide (NHPI), ananionic phase transfer catalyst (KB(C6F5)4),and a copper(I) bisimine complex.
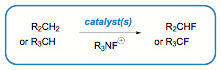
Scheme 3. Aliphatic Fluorination
We turned our attention to an investigation of the reaction's scope. A variety of aliphatic, allylic,and benzylic substrates were investigated. Unfortunately, upon initial screening, the conditions optimized for reactive adamantane yielded only trace amounts of the desired fluorinated products on a variety of substrates. At this point, we chose to focus our efforts on a less reactive model substrate such as cyclododecane. We found that heating the reaction increased the yield greatly (43%), a likely result from increased reaction rate, and as the product is a secondary fluoride, solvolysis in MeCN was not such a serious problem. In an effort to improve both the yield and the rate of reaction, N-hydroxyphthalimide 7 (10 mol%), which is known to form the "PINO" radical 8 insitu in the presence of redox active metals (eq 3), was examined as a cocatalyst. Interestingly enough, addition of NHPI provided further increases in yield of 9(63%), and along with additive KI to form the putative Cu(I) ate complex 9 (10 mol%), afforded optimal reaction (72%, Table 2).
Scheme4.
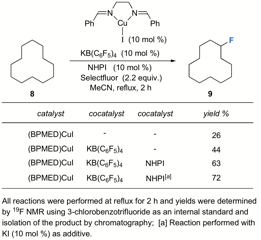
Table1.
Using the optimized conditions of Table 1, we explored other cycloalkanes, as they each give rise to one distinct monofluorinated product. As stated, cyclododecane fluorinated smoothly under the optimized conditions to afford the monofluoride in 72% yield. When longer reflux times were employed, 1,1-difluorocyclododecane began to form in appreciable amounts (18% after 24 h). Medium sized rings such ascycloheptane, cyclooctane,and cyclodecane worked as well. Given the fact that an excess of cycloalkane is not used, it is remarkable that more polyfluorination is not observed. In fact, conditions can be found under which the monofluoride is virtually the exclusive fluorinated product. On the other hand, extended reaction times for entries 3 and 5 lead to diminished yields. It is clear that the products undergo a slow solvolysis reaction, an unsurprising observation given the demonstrable release of Prelog strain during SN1 reactions of 8- and 10-membered ring systems, and that reaction times in MeCN reflect the susceptibility of substrates to solvolysis.
Straight chain substrates such as n-dodecanegive rise to a virtual 1:1:1:1:1 mixture of monofluorinated products in 63% yield, although the necessity of 1.2 equiv KI may reflect their less reactive nature (entry 10). Allylic substrates proved to be interesting in their own way. For example, a-methylstyrenes are known to fluorinate in MeCN to form fluoroacetamides(under so-called electrophilic conditions), admixed with variable quantities of allylic fluorides. Under catalytic conditions at room temperature, as demonstrated herein, the allylic fluorides predominate to the virtual exclusion of the fluoroacetamides. This would seem to bolster the case for a different (non‑electrophilic) mechanistic pathway as well.
Table3.Catalytic Substrate Fluorinations

At this point we undertook a preliminary UV-Vis study of components of the catalytic system. The main observations include: 1) Bisimine ligand (BPMED) + CuI affords aspectrum consistent with a Cu(I) complex; 2) addition of KI maintains theoxidation state of copper at (I); 3) addition of Selectfluor gives rise to weak bands indicative of Cu(II) that disappear rapidly, concomitant with the appearance of a prominent I3- band. In turn, addition of NHPI results in consumption of I3-. Presumably, Cu(I) is regenerated as well. In any case, it is clear that this is a complex system and further investigations will prove essential to the elucidation of a reaction mechanism. Additional study will address applications to complex substrates and natural products.









