Reports: DNI451435-DNI4: Theoretical and Experimental Investigation of Oxenium Ions
 printer friendly
printer friendly1. Theoretical Investigation of Phenyloxenium Ion
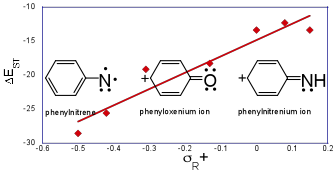
The geometries and energies of the electronic states of phenyloxenium ion (Ph-O+) were computed at the multireference CASPT2/pVTZ level of theory. Despite being isoelectronic to phenylnitrene , the phenyloxenium ion has remarkably different energetic orderings of its electronic states. The closed-shell singlet configuration (1A1) is the ground state of the phenyloxenium ion , with a computed adiabatic energy gap of 22.1 kcal/mol to the lowest energy triplet state (3A2). Open-shell singlet configurations (1A2, 1B1, 1B2, 21A1) are significantly higher in energy (> 30 kcal/mol) than the closed-shell singlet configuration. These values suggest a revision to the current assignments of the ultraviolet photoelectron spectroscopy bands for the phenoxy radical to generate the phenyloxenium ion . For para-substituted phenyloxenium ions, the adiabatic singlet-triplet energy gap (DEST) is found to have a positive linear free energy relationship (LFER) with the Hammett-like s+R/s+ substituent parameters; for meta substituents, the relationship is non-linear and negatively correlated. CASPT2 analyses of the excited states of p-amino phenyloxenium ion and p-cyano phenyloxenium ion indicate that the relative orderings of the electronic states remain largely unperturbed for these para substitutions. In contrast, meta-donor substituted phenyloxenium ions have low-energy open-shell states (open-shell singlet, triplet) due to stabilization of a p,p* diradical state by the donor substituent. However, all of the other phenyloxenium ions and larger aryloxenium ions (naphthyl, anthryl) included in this study have closed-shell singlet ground states. Consequently, ground-state reactions of phenyloxenium ions are anticipated to be more closely related to closed-shell singlet arylnitrenium ions (Ar-NH+) than their isoelectronic arylnitrene (Ar-N) counterparts.
2. Theoretical Investigation of Heteroaryl Oxenium Ions
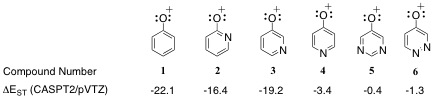
The electronic state orderings and energies of heteroaryl oxenium ions were computed using high-level CASPT2//CASSCF computations. We find that these ions have a number of diverse, low-energy configurations. Depending on the nature of the heteroaryl substituent, the lowest-energy configuration may be open-shell singlet, closed-shell singlet, or triplet, with further diversity found among the subtypes of these configurations. The 2- and 3-pyridinyl oxenium ions show small perturbations from the phenyl oxenium ion in electronic state orderings and energies, having closed-shell singlet ground states with significant gaps to an n,p* triplet state. In contrast, the 4-pyridinyl oxenium ion is computed to have a low-energy nitrenium ion-like triplet state. The pyrimidinyl oxenium ion is computed to have a near degeneracy between an open-shell singlet and triplet state, and the pyrizidinyl oxenium ion is computed to have a near triple degeneracy between a closed-shell singlet state, an open-shell singlet state, and a triplet state. Thus, the ground state of these latter heteroaryl oxenium ions cannot be predicted with certainty; in principle, reactivity from any of these states may be possible. These systems are of fundamental interest for probing the spin and configuration-dependent reactivity of unusual electronic states for this important class of reactive intermediate.
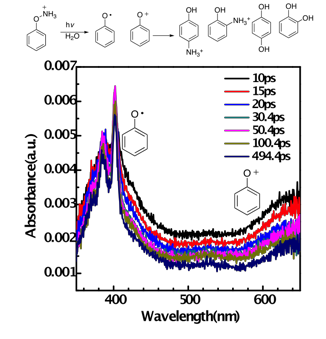
3. Experimental Generation and Reactivity of the Phenyloxenium Ion
Photolysis of protonated phenylhydroxylamine was studied using product analysis, trapping experiments, and laser flash photolysis experiments (UV-Vis and TR3 detection) ranging from the femtosecond to the microsecond timescale. We find that the singlet excited state of the photoprecursor partitions within 5 ps into two channels, generating both a longer-lived low-wavelength absorbing transient (5 ns) that we assign to the phenoxy radical and a very short-lived (80 ps) longer-wavelength absorbing transient that we assign to the phenyloxenium ion. Product studies from photolysis of this precursor show rearranged protonated o/p-aminophenols and solvent water adducts (catechol, hydroquinone). The former products can be largely ascribed to radical recombination or ion recombination, while the latter are ascribed to solvent water addition to the phenyloxenium ion. The phenyloxenium ion is apparently too short-lived under these conditions to be trapped by external nucleophiles other than solvent, giving only trace amounts of chloro and azido adducts upon addition of chloride and azide traps, respectively. Product studies upon thermolysis of this precursor give the same products as those generated from photolysis, with a difference being that the ortho adducts (o-aminophenol, o-hydroquinone) are the major products from heating (higher ratio), whereas the para adducts are the major products from photolysis.