Reports: UNI150347-UNI1: Toward Greater Understanding and Expanded Utility of the Palladium-Catalyzed Activation of Carbon-Carbon Single Bonds
 printer friendly
printer friendlyThe activation and functionalization of carbon-carbon single bonds remains an unrealized facet of organic chemistry. A generalized transition metal-catalyzed methodology allowing for the selective cleavage of carbon-carbon bonds has nearly boundless potential, yet efforts to date have primarily relied upon specialized substrates for the achievement of this unusual reaction.[1] Likewise, subsequent functionalization of the resulting organometallic species has also proven to be a significant challenge.
 To date,
there are very few examples of mechanistic investigations into transition
metal-catalyzed carbon-carbon bond activation processes. To remedy this dearth of information, our
group has pursued the mechanistic investigation of two such reactions: the
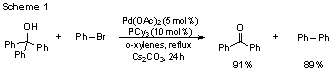
palladium-catalyzed β-aryl elimination of triarylmethanols (Scheme 1)[2] and
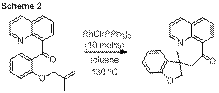
the rhodium-catalyzed alkene carboacylation using quinolinyl ketones (Scheme
2).[3] The
intent of each of these studies is to identify factors that influence the
carbon-carbon bond step of each process and subsequently utilize this
information for the extension of known reactivity.
To date,
there are very few examples of mechanistic investigations into transition
metal-catalyzed carbon-carbon bond activation processes. To remedy this dearth of information, our
group has pursued the mechanistic investigation of two such reactions: the
palladium-catalyzed β-aryl elimination of triarylmethanols (Scheme 1)[2] and
the rhodium-catalyzed alkene carboacylation using quinolinyl ketones (Scheme
2).[3] The
intent of each of these studies is to identify factors that influence the
carbon-carbon bond step of each process and subsequently utilize this
information for the extension of known reactivity.
Palladium-Catalyzed β-Aryl Elimination
 Our efforts to discern between two
mechanistic hypotheses (Scheme 3) utilized two.
Two students (one supported by a Moissan Undergraduate Fellowship
sponsored by the American Chemical Society Division of Fluorine Chemistry) were
involved in preparing a number of substituted triarylmethanols for the
identification of factors, if any, that influenced the relative propensity of
β-aryl elimination. A third student
performed reactions of a more mechanistic nature to examine the reversibility
of various steps throughout the catalytic reaction.
Our efforts to discern between two
mechanistic hypotheses (Scheme 3) utilized two.
Two students (one supported by a Moissan Undergraduate Fellowship
sponsored by the American Chemical Society Division of Fluorine Chemistry) were
involved in preparing a number of substituted triarylmethanols for the
identification of factors, if any, that influenced the relative propensity of
β-aryl elimination. A third student
performed reactions of a more mechanistic nature to examine the reversibility
of various steps throughout the catalytic reaction.
 Approximately 30 aryldiphenylmethanol
compounds containing variable substitution were prepared and subjected to the
standard reaction conditions. As
indicated in Scheme 4, several products are possible in these reactions, and
the relative ratio of these products was determined utilizing GC/MS methods
thus providing the means to examine the cleavage propensity of each
substituent.
Approximately 30 aryldiphenylmethanol
compounds containing variable substitution were prepared and subjected to the
standard reaction conditions. As
indicated in Scheme 4, several products are possible in these reactions, and
the relative ratio of these products was determined utilizing GC/MS methods
thus providing the means to examine the cleavage propensity of each
substituent.
Notably, variation of the electronics of an aryl group had only subtle influence on selectivity (Table 1), and the results demonstrated no clear trends of reactivity both more electron deficient and electron rich substituents appeared to increase the rate of β-aryl elimination versus the phenyl substituent.
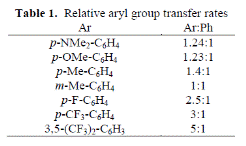 |  | ||
A clearer picture was obtained with the preparation and testing of a series of fluorine substituted substrates (Table 2). The significant influence of an ortho-fluorine is readily apparent. Beyond the ortho-substitution, additional electron withdrawing substituents result in greater selectivity, and the inclusion of multiple fluorine atoms results in cumulative effects.
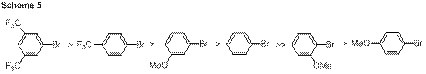
 Investigation of aryl bromides was performed
by simultaneously utilizing two aryl bromides.
The reaction was stopped short of complete conversion and the product
ratios were determined via GC/MS. Results
from these competition reactions demonstrated that electron deficient aryl
bromides proceeded more rapidly than electron rich aryl bromides (Scheme 5). These results indicate that oxidative
addition either limits the rate of catalyst turnover or lies in a reversible
step that precedes the turnover limiting step.
Investigation of aryl bromides was performed
by simultaneously utilizing two aryl bromides.
The reaction was stopped short of complete conversion and the product
ratios were determined via GC/MS. Results
from these competition reactions demonstrated that electron deficient aryl
bromides proceeded more rapidly than electron rich aryl bromides (Scheme 5). These results indicate that oxidative
addition either limits the rate of catalyst turnover or lies in a reversible
step that precedes the turnover limiting step.
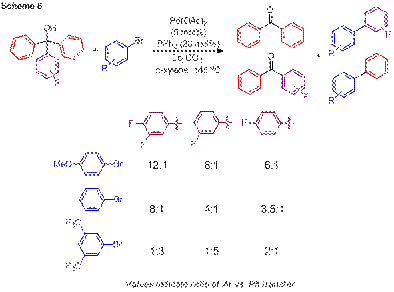
Additional information implicating initial oxidative addition has been obtained by determining the influence of β-aryl elimination. As illustrated in Scheme 6, more electron rich aryl bromides result in greater preference for cleavage of the electron deficient fluorinated aryl substituent, while the use of an electron deficient aryl bromide results in a decrease and eventual reversal in the selectivity of aryl group transfer. These results provide significant support for the premise that the selectivity of aryl transfer can be controlled by tuning the electronic character of the palladium catalyst.
To date, this work has provided significant insight into factors influencing β-aryl elimination, including the effects of electronics on the C-C activation step and the nature of the active palladium species. The results described above are currently under consideration for publication. Research efforts will continue with the expansion of this known methodology toward more widely applicable coupling methodologies utilizing carbon-carbon bond activation as an entry point into high energy organometallic species.
Rhodium-Catalyzed Carboacylation of Alkenes
Carbon-carbon single bond activation may also occur via an oxidative addition process, a process exemplified by the intramolecular carboacylation of quinolinyl ketones. In order to compare and contrast various methods of C-C bond activation, our group has also investigated the mechanism of this transformation.
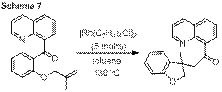 Although this project was primarily funded
via other sources, two students supported by the PRF assisted in the synthesis
and characterization of quinolinyl ketone species that were utilized in the
study. The determination of the rate
law, 12C/13C kinetic isotope effect, and rate variations
with substrates ultimately uncovered a mechanism in which the turnover limiting
step of catalysis varied with the nature of the substrate. With small alkenes, carbon-carbon bond activation
is the highest activation barrier, while larger alkenes are limited by alkene
insertion. These results have been
published.[4]
Although this project was primarily funded
via other sources, two students supported by the PRF assisted in the synthesis
and characterization of quinolinyl ketone species that were utilized in the
study. The determination of the rate
law, 12C/13C kinetic isotope effect, and rate variations
with substrates ultimately uncovered a mechanism in which the turnover limiting
step of catalysis varied with the nature of the substrate. With small alkenes, carbon-carbon bond activation
is the highest activation barrier, while larger alkenes are limited by alkene
insertion. These results have been
published.[4]
Student and PI Impact
The student involved in the various aspects of this program have combined to provide 16 oral and poster presentations to date, including presentations at the 241st National ACS Meeting in Anaheim, CA, the 42nd National Organic Symposium in Princeton, NJ, and the 243rd National ACS Meeting in San Diego, CA. Of the five students who have worked on this project, one will be attending graduate school in chemistry at the University of California-Irvine in the fall of 2012, two are rising seniors who intend to pursue graduate studies in the fall of 2013, and two are prospective dental students. All remain active in the research laboratory.
The results above have led to one publication, as well as an additional submitted manuscript. These results have also provided the foundation for additional ongoing studies into the development of methodologies for the selective activation of carbon-carbon bonds. The preliminary results obtained from this work contributed to the recent acquisition of an NSF-CAREER award that will support this work through 2017.
References
[1]) Murakami, M.; Ito, Y. Topics in Organometallic Chemistry 1999, 97-129.
[2]) Terao, Y.; Wakui, H.; Satoh, T.; Miura, M.; Nomura, M. J. Am. Chem. Soc. 2001, 123, 10407.
[3]) Dreis, A. M.; Douglas, C. J. J. Am. Chem. Soc. 2009, 131, 412.
[4]) Lutz, J. P.; Rathbun, C. M.; Stevenson, S. M.; Powell, B. M.; Boman, T. S.; Baxter, C. E. Zona, J. M.; Johnson, J. B. J. Am. Chem. Soc. 2012, 134, 715-722.









