Reports: DNI651000-DNI6: Effects of Quantum Motion of Nuclei on Reactivity of Hydrocarbons Studied in the Approximate Quantum Trajectory Framework
 printer friendly
printer friendlyQuantum-mechanical nature of nuclei, in particular of protons, is essential for understanding of chemical reactions in condensed phase, such as enzyme environment, nano-scale carbon structures or membranes. The dominant quantum-mechanical effects, present even in large molecular systems, are tunneling, internal energy flow and nonadiabatic dynamics involving excited electronic states. Our work is focused on theory development and applications of approximate quantum molecular dynamics based on the quantum trajectory formulation of the Schrödinger equation. The approximations are necessary to study molecular systems beyond a few atoms, because of exponential scaling of numerical cost of the traditional "exact" QM methods. The trajectory formulation is particularly appealing as it is well-suited for direct simulations, where the electronic structure is solved on-the-fly.
The specific goal for this grant is to study effects of zero-point-energy and tunneling on reactivity of systems of carbon and hydrogen atoms. In systems comprised of 10-100 atoms, we expect the dominant quantum effects to be associated with moving protons participating in rearrangements or bond breaking. Thus, we have started by developing a theoretical framework: the quantum trajectory formulation is implemented approximately and includes the quantum correction on dynamics into selected degrees of freedom within a single full-dimensional trajectory ensemble. The quantum correction, i. e. the so-called quantum force acting on the nuclei in addition to the classical force, is defined just for the light particles. Yet there is no global averaging over the heavy particles, due to introduction of spatial domains or subspaces dependent on positions of heavy particles. This strategy allows cheap low-dimensional estimates of the quantum correction to dynamics while allowing interactions within the wavefunction describing various channels (e. g. reactants, products) of the classical degrees of freedom.
Figure 1 illustrates the concept on a model consisting of light particle (vibrational degree of freedom, x-coordinate) coupled to a heavy particle (scattering degree of freedom, y-coordinate). The ratio of masses is 10. The quantum correction is a function of the coordinate x, but parametrically depends on the domains defined in the y-coordinate. The system is initially in the ground vibrational state; the overlaps of the time-dependent wavefunction with the ground and first excited states of the reactants and products are shown to converge as the number of domains, L, increases up to 20. The formulation has been also applied to a non-reactive collision of He and HBr, modeled in two dimensions. The conceptual challenge there was to describe the energy transfer between the two particles, while maintaining the zero-point energy in the quantum degree of freedom. Figure 2 illustrates that the concept works. However more high-dimensional studies are needed to work out a robust practical algorithm for definition of spatial domains, or perhaps subspaces in the configuration space of heavy particles, and to improve convergence properties of the correction with respect to the number of trajectories.
Our pilot study of experimentally relevant chemical systems gives estimates of the zero-point energy of CH (CD) stretch in isobutane, modeled in Cartesian space. The maximum in the spectrum of the wavepacket auto-correlation function (Figure 3) corresponds to the zero-point energy value. The wavepacket was represented with 2000 quantum trajectories that were propagated for 1000 time steps with the classical forces coming from the on-the-fly electronic structure calculations based on the Density Functional Tight Binding method. The calculation took 2 hours on a single processor; code parallelization, now in progress, will allow studies of large systems.
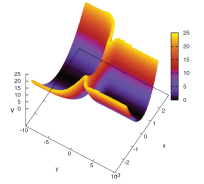
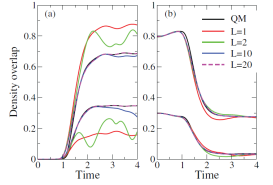
Figure 1. Left panel: the model potential; reactant/product region corresponds to negative/positive y-coordinate. Right panel: the density overlaps with the ground and excited vibrational states for the products (a) and reactants (b) obtained from dynamics with L=1, 2, 10 and 20 domains respectively shown with red, green, blue and magenta curves. The higher (lower) sets of curves show the overlap with the ground (first excited) state wavefunction density. The ground state overlaps on (b) are shifted by 0.2 in y-coordinate for clarity. Exact QM results are shown in black.


Figure 2. Nonreactive collision of He and HBr. The overlap with the ground vibrational state and average position of the quantum degree of freedom describing HBr. The results for different collision momentum Py are shifted by 0.4 for clarity. Quantum and trajectory results are shown with solid lines and dashes respectively.
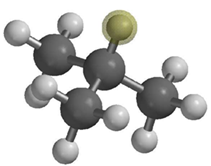

Figure 3. Estimates of zero-point energy of CH (CD) stretch in isobutane. The highlighted proton is treated as the only quantum nucleus in Nq=1 calculations. In Nq=2 calculations quantum corrections were also added for the carbon nucleus to which the quantum proton is bonded to. The vertical lines indicate the zero-point energy from an ab initio electronic structure calculation.
The formal theory development is largely finished and published (though new issues are likely to come up as we move to applications). A postdoc, who joined the group in August of 2012 will use this methodology to study carbon/hydrogen systems.
In addition to the project described above, the grant was used to support the following related research:
A graduate student in the group examined quasi-classical and approximate quantum trajectory dynamics for several model systems (H+O2 and Hennon-Heiles two-dimensional systems). Preliminary results from this work show: (i) in the quantum trajectory formulation the momentum-spreading in the scattering degree of freedom gives accurate reaction probabilities for collision energies higher than the barrier top; (ii) stabilization of the approximate quantum trajectory dynamics is needed to describe zero-point energy in the anharmonic vibrational potentials.
A postdoc who spent three months in our group and had to leave because of health problems, performed quasi-classical trajectory calculations for the O+C2H2 reaction on the parametrized surface of S. Braams (unpublished). Preliminary results have shown that the dominant reaction channel is H+HCCO and H+CH+CO (the threshold to the reactions is about 0.7 eV) in agreement with experimental observations. The other theoretically observed channel is CH2+CO. More studies are required to validate the surface and obtain simulations relevant to experiments from the experimental group of T. Minton (Montana State University).









