Reports: ND350971-ND3: Electrocatalytic Water Oxidation by Manganese Pyridinophane Complexes
 printer friendly
printer friendly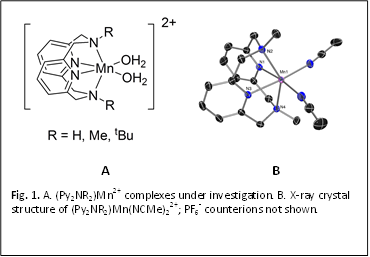 Metallation
of the pyridinophane ligands is readily achieved by reaction of the appropriate
macrocycle with MnBr2. The resulting colorless (Py2NR2)MnBr2
(R = H, Me, tBu) complexes have good solubility and stability over a
wide pH range. Conductivity measurements reveal the
Metallation
of the pyridinophane ligands is readily achieved by reaction of the appropriate
macrocycle with MnBr2. The resulting colorless (Py2NR2)MnBr2
(R = H, Me, tBu) complexes have good solubility and stability over a
wide pH range. Conductivity measurements reveal the ![Fig. 2. X-ray crystal structure of [(Py2NH2)MnIII(m-O)2MnIV(Py2NH2)]3+; counterions not shown.](images/abimages/Paper_11846_abstract_16838_0.png) complexes
to be fully ionized in neutral aqueous solution.
complexes
to be fully ionized in neutral aqueous solution.
In the solid state, all the complexes are air stable, however in aqueous solution, the complex with R = H shows a slight change in color over the course of a few days, possibly due to aerobic oxidation. Anion metathesis with nitrate increases the rate of coloration, eventually forming the green complex [(Py2NH2)MnIII(m-O)2MnIV(Py2NH2)]3+, which has been characterized by X-ray crystallography (Fig. 2). This complex has similar structural features as reported [MnIII(m-O)2MnIV] dimers. The same air sensitivity is not observed for R = Me, tBu, likely because the bulkier R groups prevent the formation of the [Mn(m-O)2Mn] diamond core.
 2.
H2O2 disproportionation
2.
H2O2 disproportionation
We have investigated the manganese pyridinophane complexes as
hydrogen peroxide disproportionation catalysts (i.e. catalase mimics) due to
the potential relationship between catalase and water oxidation reactivity. The
complexes with R = H, Me are found to be good hydrogen peroxide disproportionation
catalysts in aqueous solution. There is no catalase reactivity when R = tBu.
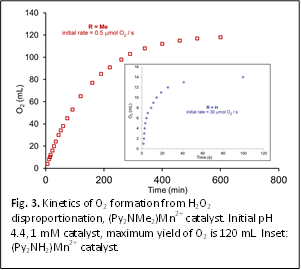
Preliminary kinetics investigations using volumetric measurements of O2
formation reveal that when R = H, the complex has moderate activity, but
limited robustness, having a turnover number (TON) of 830 at pH 4 (Fig. 3,
inset). The complex is active under acidic and basic conditions, with the
greatest activity and longevity at pH = 9 (initial rate = 40 mmol O2 /s, TON 1940). When R = Me,
the catalyst is more robust (TON > 66000), albeit slower (Fig. 3). The activity
of this particular catalyst is remarkable because: (1) the TON is several
orders of magnitude greater than any other reported catalase mimics; and (2) it
is active in aqueous solution, conditions under which most catalase mimics are
not stable. We are currently undertaking a more comprehensive investigation of
the kinetic behavior of these complexes to more fully delineate their catalase
activity, as well as other likely oxidative behavior (e.g. peroxidase
reactivity). This will allow for better comparison with previously reported
catalase mimics.
 3.
Electrochemical water oxidation
3.
Electrochemical water oxidation
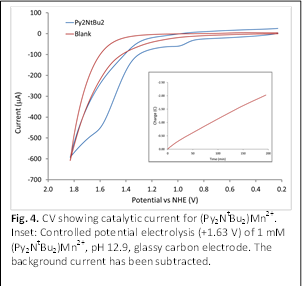
The cyclic voltammograms of (Py2NR2)Mn2+
(R = Me, tBu) in basic buffer (pH 12.9) both reveal catalytic waves with
an overpotential for water oxidation of ca. 830 mV (Fig. 4). Bulk
electrolysis experiments provide further evidence for catalysis, with good
stability when R = tBu (Fig. 4, inset). The formation of gas is
observed at the working electrode is observed. Control experiments to verify
the homogeneous catalytic water oxidation reactivity of this complex are in progress.
Our working hypothesis is that the bulkier tBu substituents prevent
the formation of dead-end [Mn(m-O)2Mn]
diamond core structures and divert the reactivity away from H2O2
disproportionation towards water oxidation. Mechanistic and computational
studies to better understand the reaction mechanism will be conducted in the
upcoming year.
4. Electrochemical water reduction
 Although
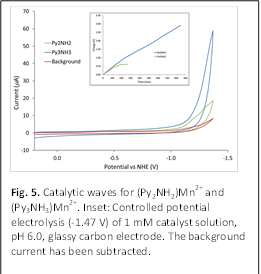
(Py2NH2)Mn2+ does not catalyze water
oxidation, we were pleasantly surprised to observe a catalytic wave with an
overpotential of ca. 1.4 V for water reduction (Fig. 5). Bulk
electrolysis (pH 6) reveals the complex to be unstable under these conditions,
with complete loss of activity within 3 h of electrolysis (Fig. 5, inset).
Despite this setback, this is a very exciting result as we are unaware of any
reports of water reduction by a manganese complex under aqueous conditions. The
other pyridinophane complexes, (Py2NR2)Mn2+ (R
= Me, tBu) decompose with the deposition of metallic manganese under
the same conditions. The macrocycle N-H protons are therefore critical to the
observed reactivity, possibly acting as proton donors.
Although
(Py2NH2)Mn2+ does not catalyze water
oxidation, we were pleasantly surprised to observe a catalytic wave with an
overpotential of ca. 1.4 V for water reduction (Fig. 5). Bulk
electrolysis (pH 6) reveals the complex to be unstable under these conditions,
with complete loss of activity within 3 h of electrolysis (Fig. 5, inset).
Despite this setback, this is a very exciting result as we are unaware of any
reports of water reduction by a manganese complex under aqueous conditions. The
other pyridinophane complexes, (Py2NR2)Mn2+ (R
= Me, tBu) decompose with the deposition of metallic manganese under
the same conditions. The macrocycle N-H protons are therefore critical to the
observed reactivity, possibly acting as proton donors.
Since it is likely that only one coordination site is
necessary for proton reduction, we decided to investigate the water reduction
reactivity of the 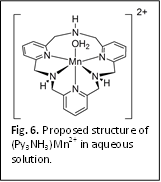 related
complex, (Py3NH3)Mn2+. The macrocycle in this
complex is proposed to act as a pentadentate ligand (Fig. 6). This binding mode
provides a free amine donor with sufficient flexibility to act as a proton
donor. Gratifyingly, this complex has a larger catalytic current at slightly lower
overpotential (Fig. 5) and substantially greater stability under catalytic
conditions (Fig. 5, inset). The formation of a gas at the working is electrode
is also observed during bulk electrolysis, which we are in the process of detecting
and quantifying. We plan to extend this investigation to other metals that are
easier to reduce.
related
complex, (Py3NH3)Mn2+. The macrocycle in this
complex is proposed to act as a pentadentate ligand (Fig. 6). This binding mode
provides a free amine donor with sufficient flexibility to act as a proton
donor. Gratifyingly, this complex has a larger catalytic current at slightly lower
overpotential (Fig. 5) and substantially greater stability under catalytic
conditions (Fig. 5, inset). The formation of a gas at the working is electrode
is also observed during bulk electrolysis, which we are in the process of detecting
and quantifying. We plan to extend this investigation to other metals that are
easier to reduce.
5. Career Impact and Student Impact
The PI is a member of a team from three campuses in the state (NM Tech, NMSU and UNM) that is developing methods for the use of solar fuels. The results from the ACS-PRF supported project have been used as preliminary data as part of a solar fuels sub-project ($2 million) of the New Mexico EPSCoR renewal application.
A graduate student is currently employed on the project, beginning August 2012, and will continue to be employed for the upcoming year. This research assistant support will allow him to devote more time to the project.









