Reports: UR150719-UR1: Regioselective Semihydrogenation of Dienes
 printer friendly
printer friendlyPrior to our work on this project, there existed no general strategy for the direct, regioselective semihydrogenation of dienes that is selective for the more highly substituted alkene of a differentially substituted diene. Our project proposal was based on promising preliminary results indicating that a one-pot, reversible hydroboration strategy would be effective for simple diene substrates and had the following five objectives:
(1) Determining the Functional Group Tolerance of the Regioselective Semihydrogenation Methodology
(2) Determining the Substrate Scope of the Regioselective Semihydrogenation Methodology
(3) Expanding the Methodology to Include Chemoselective Hydrogenolysis
(4) Adapting the Methodology into a General Strategy for the Isomerization of Stilbenes
(5) Expanding the Synthetic Potential of Ring-Closing Enyne Metathesis (RCEM)
During the first grant year of this project, we achieved Objectives 1 and 4 and partially achieved Objective 2. This has resulted in two publications. Completing Objectives 2 and 3 is our goal for the upcoming year of the grant.
Objectives 1 and 2
9-BBN-H was selected as an alkene protective group for three principal reasons: (1) monohydroboration of differentially substituted terminal dienes is exceptionally regioselective for the terminal alkene when bulky dialkylboranes are used, leaving the more highly substituted alkene intact, (2) trialkylboranes are hydrogenated under heterogeneous catalysis only at elevated temperatures and pressures, and (3) trialkylboranes can be cleaved at elevated temperatures or in the presence of certain electrophiles, revealing the original alkene and producing an oxidized borane. This suggests a three-step hydroboration, hydrogenation, oxidation sequence to accomplish the desired regioselective transformation.
Since the first two steps of this sequence occur without production of stoichiometric byproduct, we envisioned a one-pot transformation involving sequential addition of 9-BBN-H, followed by hydrogen and catalytic Pd/C, and finally an electrophile. We evaluated a number of commercially available electrophiles and selected the powerful electrophile 2-methyl-2-nitrosopropane (MNP) because it oxidized the intermediate trialkylboranes quantitatively and within minutes to reveal the initial alkene. If ethanolamine is added following the final oxidation step, boron-ethanolamine complexes precipitate out of solution. Thus, purification frequently requires no more than filtration through a plug of silica gel to remove Pd/C and insoluble boronates, the only reaction byproduct. This one-pot procedure leads to the regioselective semihydrogenation of a variety of dienes.
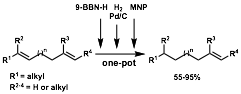
Regioselectivity is high; generally only one product alkene regioisomer is observed by NMR. Synthetically useful yields are obtained for a variety of substrates including cyclic and acyclic terminal dienes, dienes with various substitution patterns, and strained internal dienes. Importantly, a conjugated terminal diene was semihydrogenated without constitutional isomerization of the remaining olefin, which is usually observed when such reductions are attempted with homogeneous and hetereogeneous catalysts.
We also demonstrated that the third step of the sequence (dehydroboration) can be replaced by a more traditional oxidation step to yield regioselectively semihydrogenated product alcohols. This work was published in The Journal of Organic Chemistry.
Objective 4
Stilbenes and their substituted derivatives, stilbenoids, have attracted significant attention for their wide range of useful properties, which include applications in optics, biochemistry, and chemotherapy. Generally the two alkene stereoisomers of a particular E/Z stilbenoid pair exhibit meaningfully different physiochemical properties. Accordingly, methods for their stereoselective chemical synthesis and derivatization have been intensively investigated, along with strategies for the interconversion of the E and Z stereoisomers.
The most widely utilized strategy for stilbenoid synthesis is the Wittig reaction along with its more modern variants, especially the Horner–Wadsworth–Emmons reaction. A mixture of E and Z stereoisomers is generally formed, though optimization of conditions can lead to highly selective formation of the E stereoisomer. Comparatively fewer methods have been developed for the selective production of the more thermodynamically stable E stilbenoid stereoisomers via isomerization of E/Z mixtures. Recently, mild and practical palladium-catalyzed methods have been developed, but these reactions are not effective when electron-withdrawing substituents are present. Collectively, these examples illustrate that additional methods for stilbenoid isomerization are still needed.
We envisioned a one-pot, E-selective stilbenoid isomerization sequence that takes advantage of the reversibility of hydroboration in the presence of strongly oxidizing electrophiles. A mixture of E and Z stilbene isomers would undergo syn addition of 9-BBN to yield a trialkylborane. The mechanistic reverse of this addition, a syn elimination, must then occur from an eclipsed conformer, which would magnify the destabilizing effects of steric interactions on the conformational equilibrium. Syn elimination of 9-BBN with MNP from the least crowded eclipsed conformer should then regenerate the stilbenoid with high E stereoselectivity. Elimination on the borane reagent itself is precluded with 9-BBN as it would lead to the generation of a bridgehead alkene, making 9-BBN a particularly good choice of dialkylborane for this methodology.
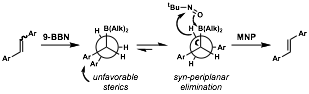
We then subjected a range of substrates to the isomerization sequence and observed very high levels of E selectivity (usually >20:1). The isomerization is tolerant of both electron-donating and electron-withdrawing substituents at various positions on the aromatic rings. We did not evaluate substituents that are reduced in the presence of dialkylboranes because a nitro group, which is relatively stable to dialkylboranes at lower temperatures, was competitively reduced to an amine at the elevated temperatures used in this study. To illustrate this methodology, we executed a short synthesis of resveratrol. This work was published in Tetrahedron Letters.
Impact of the Work
Two of the five papers that I published pre-tenure were directly funded by this grant, and I consider ACS-PRF funding to have been an essential component of my successful tenure application. So far, two undergraduates have been funded through this grant. One is applying to graduate school in organic chemistry this year, and the other is applying to medical school.









