AmericanChemicalSociety.com
Reports: G3 48059-G3: Aqueous Metal-Ligand Bifunctional Hydrogenation of Carbon Dioxide
Our group has been studying the metal-ligand bifunctional hydrogenation reaction which involves the transfer of proton form ligand and hydride from metal in the hydrogenation of polar double bonds, such as ketones and imines (Figure 1). Under the grant provided by the ACS PRF we undertook a study on the hydrogenation of carbon dioxide to formic acid using (h6-arene)Ru(II) type catalysts. In order to better understand the mechanism of carbon dioxide hydrogenation we began our investigation by examining the microscopic reverse of this reaction which is the dehydrogenation of formic acid.

Although the mechanistic details of the bifunctional transfer hydrogenation
using 2-propanol as the transfer agent have been well studied, there are less
quantitative studies on the hydrogenation in water with formic acid or the 5:2 formic
acid/triethylamine azeotropic mixture (TEAF). To gain insight into the mechanism of the
transfer hydrogenation of acetophenone in these media we employed a series of N-(p-X-phenyl)-N'-(p-toluenesulfonyl)1,2-ethylenediamine
ligands to perform structure-activity studies in TEAF. The syntheses of this ligand series was not
trivial for all substituents and lead us to develop an improved synthetic
method. The catalytic reactions were typically performed by premixing the
appropriate ligand with the ruthenium precursor [Ru(h6-p-cymene)Cl2]2
plus a base prior to addition of acetophenone (t=0 of the experiments). Reactions with independently synthesized [Ru(h6-p-cymene)(L)Cl] [L = para-substituted ligands] gave
results that were similar to in situ
catalyst preparation experiments. The
Hammett plot, based on initial rate kinetics, is shown in Figure 2a. We performed a similar Hammett study in the
2-propanol, Figure 2b. In both cases, reaction
rates with electron-withdrawing substituents were very slow and the data is not
included.
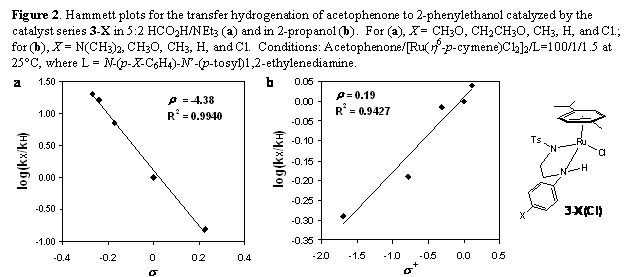
The hydrogenations of acetophenone in TEAF produced a surprisingly large
reaction constant, r = -4.38, with reasonable linear
correlation. Hammett s
values are a quantitative measure of the charge on reaction center. A r of -4.38 indicates the accumulation of
substantial amount of positive charge on the p-X-phenyl-nitrogen atom in the transition state. This result suggests that the interaction of
acetophenone with 1-X in a closed
transition state, such as that proposed for transfer hydrogenation with
2-propanol, is not plausible for this reaction.
However, an explanation that is consistent the observation of a large
and negative r is the stepwise
rate determining reaction of formic acid with 2-X to regenerate the RuH-amine 1-X.
The transition state for proton transfer to the amido ligand is shown in
Figure 3. The intermediate in the
stepwise reaction may be an ion pair, such as 4-X. Such mechanisms have
been hypothesized but not demonstrated unambiguously. Our results provide quantitative evidence
that the metal-ligand bifunctional transfer hydrogenation of acetophenone in
TEAF involves an ionic mechanism that differs from 2-propanol reactions.


In contrast, we found a positive r of 0.19 for the transfer hydrogenation in 2-propanol. A small r value is consistent with a concerted mechanism where minimal charge development is expected on any atom. The weak slope in the plot may indicate an asynchronous concerted transition state where the Ru-H bond is elongated relative to the N-H, as in Figure 4. This would result in a modest degree of bond polarity in the transition state and minimal positive charge localization on the ligand N. These results are consistent with Noyori's calculations on the transition state for the dehydrogenation of methanol by the Ru-amide complexes where the movement of methanol proton to ligand nitrogen precedes to a small degree the transfer of C-H to ruthenium. In the transfer hydrogenation of acetophenone it had experimentally been determined that the dehydrogenation of 2-propoanol was rate determining under saturating acetone conditions. It is curious that a reasonable linear free energy correlation only existed when we plotted our data against the Hammett-Brown s+ substituent constants (correlations with s ΄, s-, s*, and sI gave scattered plots). The s+ substrate constants are based on the ionization of cumyl chlorides where a significant amount of conjugation exists between the para-substituent and the cationic reaction center. This is may be evidence for the dominance of substituent resonance effects in outer-sphere ligand participation ion the hydrogenation.
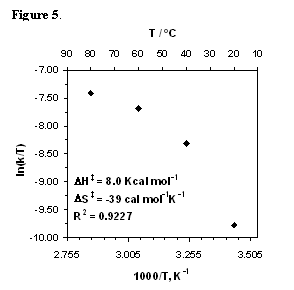
In a related study, we determined the activation parameters for the catalytic transfer hydrogenation of acetophenone in aqueous formate solution using initial rates kinetics and measuring acetophenone and 2-phenylethanol concentrations by their 1H NMR (Figure 5). The large and negative activation entropy seems more consistent with an ordered transition state, such as would be found in a concerted process. This interpretation would make the aqueous reaction resemble the 2-propanaol reactions more than the TEAF reaction. The plot in Figure 5 clearly shows curvature and a poor linear correlation, so the activation parameters need to be qualified. Downward curvature in an Eyring plot can be interpreted in terms of a composite rate constant where the rate limiting step is coupled with a prior equilibrium event; in this case, one that favors the product of the equilibrium (or the species prior to the activated rate step). Therefore, increasing temperatures would deplete the concentration of the active species and give rise to a lower than expected rates. The curvature can also indicate the existence of an activation heat capacity (DCp=¶(DHp)/¶T). Differences in ground state and transition state heat capacities may reflect differing degree of solvation or substantial solvent reorganization. The involvement of solvent molecules in the transition states for reactions in methanol have been proposed based on computational studies. These effects are expected to be more pronounced in the aqueous medium and will require further investigations.