AmericanChemicalSociety.com
Reports: DNI1 49072-DNI1: Ligand-Linked Catalysis: Metal-Catalyzed Functionalization via Transient Directing Group Installation
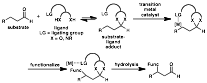
Introduction. Through the invention of synthetic reactions differential from currently existing technologies, we may further enable novel, highly efficient approaches to desired molecular structures. Our funded proposal outlined a general approach to the site-selective functionalization of relatively unreactive bonds by transition metal catalysts. This proposal represented a unification of two distinct concepts: the metal-catalyzed functionalization of unreactive bonds, and the reversible installation of a ligating species that can render a process intramolecular. In this approach, the ligand will act as the link between substrate and catalyst that allows and directs the desired reaction to occur (Figure 1). It is envisioned that an equilibrating transacetalization process between the substrate and the organic molecule catalyst will form an intermediate in situ. This covalently attached substrate-ligand adduct will then direct (via a ligating group LG) the transition metal to functionalize the substrate in a specific fashion, which upon hydrolytic release of the organic catalyst will afford the product. It is anticipated that this versatile strategy to functionalization will lead to a variety of transformations currently unrealized by either transition metal catalysis or organocatalysis approaches. Figure 1. Metal-Catalyzed Functionalization of Aldehyde-Based Substrates via Transient Covalent Attachment.
Preliminary progress. Thoughtful experimental
design and execution will be necessary in order to achieve the processes we
envision. Specifically, we will
need to evaluate the following: A) the molecular architecture of the ligand and
its intrinsic directing group capability, both spatially and electronically, B)
the transacetalization component and its inherent ability to facilitate
exchange processes with organic substrates, and C) the compatibility and
synchronization of these two aspects.
To start systematically addressing these issues, we have approached this
problem by generating our putative linked intermediates stoichiometrically, and
studied their capacity in remote functionalizations. To that end, we chose to
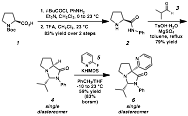
utilize a proline-based scaffold for our ligand as a starting point for these
investigations (Scheme 1). Ligands
based on the amino amide acetalization fragment showed significant
promise. N-t-Boc-proline can be
converted to 2 in two steps in good
yield. This amino amide can be
condensed with isobutyraldehyde to form 4. This compound exists as a single
diastereomer, dictated by the isopropyl group preferentially residing on the
convex face of the 5,5-ring system to avoid steric interactions. The directing group pyridyl ring is
then installed via enolization and diastereoselective alkylation with
2-fluoropyridine. Scheme 1
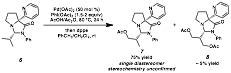
With 6 in hand, we evaluated this molecule in a directed oxidative
transformation (Scheme 2). When
aminal 6 is treated with catalytic
Pd(OAc)2 and PhI(OAc)2 as the stoichiometric oxidant,
according to the protocol of Sanford, acetate 7 is predominantly formed.
This preliminary result is exceptionally significant for several
reasons. First and foremost, sp3-hybridized C-H bond
functionalization occurs, highlighting the compatibility of this substrate with
the reaction conditions. Moreover,
the oxidation occurs with nearly singular site-selectivity. Although there are a number of C-H
bonds that could be oxidized under these conditions, the molecular framework directs
the functionalization to almost exclusively one carbon, indicating that this
oxidation process is highly diastereoselective. Scheme 2
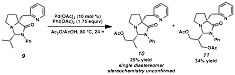
As part of developing our
understanding of the ligand framework and its ability to direct
functionalization, we have also synthesized 9 for oxidative study (Scheme 3). This compound features an additional methylene group between
the directing pyridyl and the pyrrolidine ring, which we anticipated would
impart increased conformational flexibility. This compound, when treated with the same oxidative
conditions, afforded 10 and 11, the products of mono- and
diacetoxylation respectively.
Importantly, this reaction displayed much greater reactivity relative to
the oxidation of 6. Current efforts are directed toward
further modifications on this framework, in anticipation of understanding the
effects of substitution on overall reactivity. Scheme 3
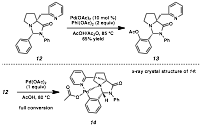
In addition to the
functionalization of sp3-hybridized
C-H bonds, we are also investigating sp2-hybridized
C-H bonds. The same ligand
framework of 9 can be attached to
benzaldehyde to form 12 (Scheme 4). Again using palladium oxidative
catalysis, acetate 13 was
formed. We were also able to
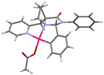
isolate a cyclometallated complex by treating 12 with stoichiometric Pd(OAc)2, and obtain a crystal
structure of the complex. As can
be seen in this structure, both the pyridyl and the pyrrolidine nitrogen atoms
are bound to the metal center, and they appropriately position the metal to
engage the aromatic ring. It
should also be noted that the aromatic ring is on the convex face of the
bicyclic system defined by the amino amide-based aminal, consistent with our
scaffold design. Scheme 4
Scheme 5
Summary. We anticipate that this type
of transformation, once fully developed, could be widely applicable in
oxidative functionalization reactions.
We plan to investigate the potential of this reactivity in a number of
different oxidative manifolds. We
also anticipate this concept to be applicable in hydroarylation and hydrovinylation
reactions, as well as in C-C bond insertion processes. Our current plan is to publish our
aforementioned efforts in the oxidative functionalization as a communication,
and then further explore the reactivity in these different arenas. The innovative nature of this work is
likely to garner attention in the organic and organometallic chemistry
communities. I have presented
posters detailing our efforts at two Gordon Conferences and a DOE contractors
meeting that have been very well-received. My graduate student Erin Stache, who has been the main
contributor to these efforts, had an incredibly successful second-year oral
examination, which speaks to her progressing development as a scientist due to
her participation in this project.
Funding of this proposal has been instrumental in the launching of this
project.
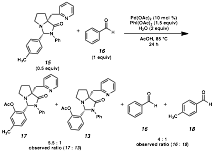
 Lastly, we have started to
see evidence of synchronized oxidation with ligand exchangability. We have treated aminal 15, derived from p-tolualdehyde, and benzaldehyde under the oxidative conditions
with added water to assist in exchange (Scheme 5). Although 17 is
the predominant oxidative product, we have observed small amounts of 13, arising from the ligand framework
hydrolyzing off tolualdehyde, condensing with benzaldehyde, and then directing
oxidative functionalization. We
anticipate that further evaluation of substrate and conditions will lead to a
fully optimized process, and open the door for much greater generality in these
types of transformations.
Lastly, we have started to
see evidence of synchronized oxidation with ligand exchangability. We have treated aminal 15, derived from p-tolualdehyde, and benzaldehyde under the oxidative conditions
with added water to assist in exchange (Scheme 5). Although 17 is
the predominant oxidative product, we have observed small amounts of 13, arising from the ligand framework
hydrolyzing off tolualdehyde, condensing with benzaldehyde, and then directing
oxidative functionalization. We
anticipate that further evaluation of substrate and conditions will lead to a
fully optimized process, and open the door for much greater generality in these
types of transformations.