AmericanChemicalSociety.com
Reports: AC6 48623-AC6: Multi-Scale Modeling of Reaction with Diffusion
The goals of this project are to achieve fundamental understanding of the influence of confinement in nano-pores on diffusion and chemical reaction, using theory and simulation at both the electronic and atomistic scales of matter.
We have carried out molecular dynamics simulations for nano-porous carbons to study the effects of pore morphology and topology on self-diffusion. For single walled carbon nanotubes of increasing pore diameter we find a cross-over from single file diffusion (one dimensional random walk) to much faster Fickian diffusion (three dimensional random walk) for pore diameters of 2.8s, where s is the diameter of the diffusing molecule [1,2]. Many catalyst materials, as well as carbon electrodes used in fuel cells and supercapacitors, have a range of pore sizes that include both micropores (D<2 nm) and mesopores (2<D<50 nm), and both single file and Fickian diffusion are observed in different regions of the same material. We have used molecular dynamics simulations to study the diffusion of argon in two such model systems [2], a bundle of single walled (25,0) carbon nanotubes [3], and a widely used industrial activated carbon, BPL [2]. In both cases we observe a bimodal diffusion mechanism with molecules exhibiting single file in small pores, and Fickian diffusion in the larger pores. In Figure 1 we show snapshots of argon in a model of BPL constructed by the Hybrid Reverse Monte Carlo method; grey lines are bonds connecting carbon atoms, green spheres are argon atoms. At low pressures (left image) argon is preferentially adsorbed in the small pores, where anomalous single file diffusion occurs. At higher pressures (right image) the small pores are filled and argon is also adsorbed in the larger pores, so that diffusion is dominated by the Fickian mechanism.
Figure 1. Simulation snapshots of argon diffusing in an
industrial disordered nano-porous carbon, BPL, at 120
K at P/P0=6x10-5
(left) and P/P0=1 (right),
where P is the bulk gas pressure and P0 is the vapor pressure of
argon.
We
have also performed ab initio density functional theory
studies of the dissociation of water to generate hydrogen on titanium-decorated
buckyballs, followed by storage of the generated
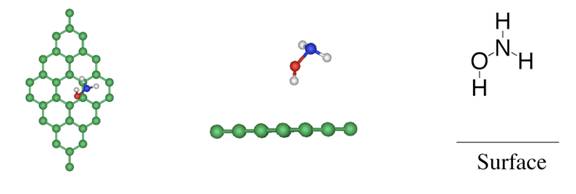
hydrogen [4], and of the dissociation of ammonia over functional groups on
graphite oxide. Ammonia is found to dissociate over carboxyl and epoxy groups
of graphite oxide [5]. The interactions
between water and functional groups at graphite oxide have also been investigated.
The results show that water does not dissociate but instead forms a hydrogen
bond with functional groups. Further examination reveals that water is preferentially
adsorbed and prevents ammonia reacting with the functional groups, in agreement
with recent experimental results.
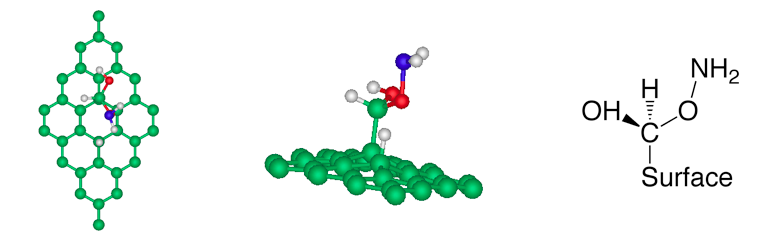
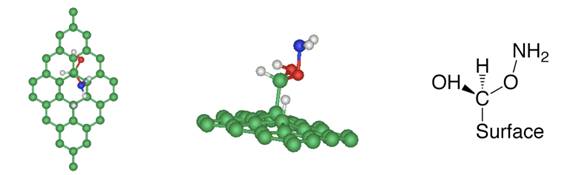
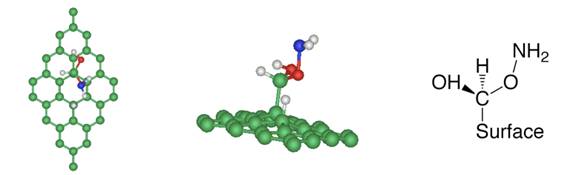
(a) NH3 + carboxyl group (b) NH3 + epoxy group Figure 2
(a) After ammonia dissociation over a COOH group of graphite
oxide, the C=O bond from the carboxyl group is broken to form new C-H and
C-O-NH2 bonds. (b) One ammonia reacts with the epoxy group of graphite
oxide, generating one hydroxylamine molecule, which is released from the
graphite oxide surface.