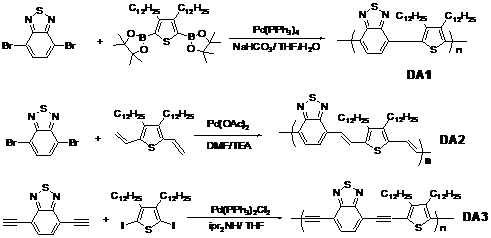AmericanChemicalSociety.com
Reports: DNI10 48733-DNI10: Demonstrate the Feasibility of Using Triplet Polymers with Variable Bandgaps for Efficient Photovoltaics
Abstract
In order to study the relationship between the bandgaps and linking bonds, three kinds of model polymers (DA1, DA2 and DA3) consisting of electron-rich (D) and electron-deficient units (A) were synthesized. They had the same electron-rich and electron-deficient groups, but were linked together by aryl-aryl (DA1), aryl-vinyl-aryl (DA2), and aryl-ethynyl-aryl (DA3), respectively. This result indicated that changing the linkers between the electron-rich and electron-deficient units can tune the HOMO energy levels of the copolymers.
a. The optical and electrochemical properties results show that, compared to the aryl-aryl and aryl-vinyl-aryl based polymers, the aryl-vinyl-aryl structure can keep almost the same LUMO energy level and increase the HOMO energy level of copolymer. Therefore, it led to the lowest bandgap of copolymer.
b. The quantum-chemical calculations results show that DA2 and DA3 have a higher degree of co-planarity (no larger than 5° dihedral angle), which may improve carrier mobility of solid film.
1. Experimental section
According to the synthesis route shown in Figure 1, three kinds of model polymers (DA1, DA2 and DA3) consisting of electron-rich (D) and electron-deficient units (A), were synthesized via Suzuki, Stille or Sonogashira polycondensation, respectively. They had the same electron-rich and electron-deficient units (disubstituted 3,4-dialkyl thiophene monomers and benzothiadiazole monomers), but were linked together by aryl-aryl, aryl-vinyl-aryl, and aryl-ethynyl-aryl respectively.
Figure
1. Chemical structures and synthesis route of copolymers with different bonds.
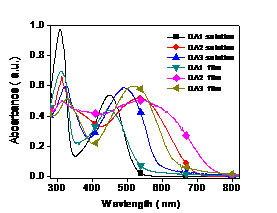
2. Optical
and Electrochemical Properties. The UV-vis absorption spectra of the three
copolymers in solution and on solid films are shown in Figure 2. The polymers' bandgaps are in the following
order: DA1 (2.22 eV) > DA3 (1.91eV) > DA2 (1.65 eV). Comparing the solution and solid film
absorption, the DA2 was red-shifted by 62nm, whereas the DA1 was 10nm and DA3 was
50nm, indicating the DA2 unit promotes better interchain or intermolecular
interaction in solid films. DA2 has the strongest red-shifted and the lowest
bandgap, which is likely due to a better p-p stacking of the polymer
backbone in the solid state.
Figure 2. UV-vis absorption spectra of DA1, DA2 and DA3 in
dilute chloroform solutions and on solid film (spin-coating from chloroform
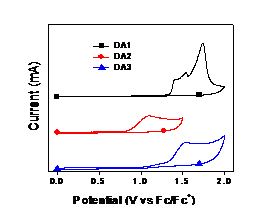
solution). The HOMO energy levels were determined from cyclic
voltammetry, and LUMO energy levels were roughly estimated from the difference
between HOMO and the optical bandgap (Eg) of the polymer films. Interestingly
they exhibited almost the same LUMO energy levels, but their HOMO energy levels
were very different (Table 1). Thus, they show different bandgaps. DA2 was
found to have the highest HOMO energy level. This result indicated that
changing the linkers between the electron-rich and electron-deficient units can
tune the HOMO energy levels of the copolymers.
Figure 3. Cyclic
voltammetry of the copolymers DA1, DA2 and DA3 in 0.05M TBAPF6 acetronitrile. 3. Quantum-chemical Calculations. All
the calculations were performed with Gaussian03 program, using the density
functional theory (DFT) at B2LYP/6-31G (d, p) levels. Figure 4 shows the side-view and front-view
of a single chain oligomer backbone structure.
The optimized structure and geometrical parameters of oligomers show
that DA1 had a dihedral angle (about 125o) between the thiophene and
thiadiazole units, which would reduce the delocalization in the backbone. DA2 and
DA3 exhibited a small dihedral angle (no larger than 5o), which
might has little effect on the delocalization on the coplanar backbone
structure.
DA1
DA2
DA3
Figure 4. Side-view
(left) and front-view (right) configuration of the copolymer single chain
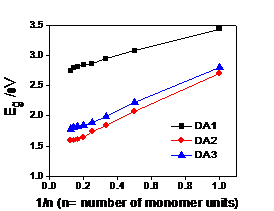
calculated from density functional theory (DFT) at B2LYP/6-31G (d, p). As shown in Table 1, the bandgaps of DA2 and DA3 are
in good agreement between the theoretically predicted values and experimentally
optical bandgap (Eg) values. However, the bandgap of calculated
results for DA1 (2.74 eV) is somehow different from the experimental value
(2.22 eV). Regarding to the order of bandgaps, the DFT calculation results were
found to be in good agreement with the experimental results: DA1>DA3>DA2.
This indicates that the linkers between the electron-rich and
electron-deficient units can tune the bandgaps of copolymers, of which the aryl-vinyl-aryl structure achieved the lowest bandgap. Table 1. HOMOs, LUMOs, and bandgaps of DA1, DA2 and
DA3 from the experiments and calculation by DFT B3LYP/6-31G (d,p).
DA1 DA2 DA3 HOMO LUMO Eg HOMO LUMO Eg HOMO LUMO Eg calculated (n=8) -5.368 -2.628 2.74 -4.544 -2.954 1.59 -4.902 -3.117 1.785 experimental result -5.69 -3.39 2.22 -5.15 -3.49 1.65 -5.43 -3.49 1.91
Figure 5. The bandgaps of DA1, DA2, and DA3 calculated by DFT B3LYP/6-31G (d,p). Conclusion Three kinds of model copolymers consisting of the same electron-rich
and electron-deficient units, but linked with different chemical bonds, were
synthesized via Suzuki, Stille or Sonogashira polycondensation, respectively. Both
the DFT calculation and experimentally obtained results show that the linking
groups between the electron-rich and electron-deficient units affect the
overall copolymer energy levels (LUMO and HOMO) and bandgaps. The aryl-vinyl-aryl (D-vinyl-A) structure
showed the greatest potential for light harvesting in organic solar cells.
Acknowledgement Acknowledgment is made to Donors of the American Chemical
Society Petroleum Research Fund for support of this research. ADDIN EN.REFLIST