AmericanChemicalSociety.com
Reports: GB3 47598-GB3: Chelation Directed C-H Functionalization Reactions with Ruthenium Boryl Complexes
Directed C-H functionalization (carbon-hydrogen bond to carbon-heteroatom bond) is becoming a valuable tool in organic synthesis. Applications of this methodology, however, are currently limited by the carbon-heteroatom bond that can be formed. C-H functionalization with metal boryl complexes provides regioselective formation of carbon-boron bonds (selective for aryl and terminal alkyl C-H bonds) from carbon-hydrogen bonds. Because the C-B bond can be converted into C-C, C-O, C-N, and other C-X bonds, this method provides a very general way to convert simple substrates into synthetically valuable products if a single reactive site is present in the substrate (for example, one terminal –CH3 group).
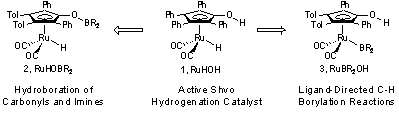
This report describes our progress in the synthesis and evaluation of the reactivity of boron-substituted analogues of metal–ligand bifunctional catalysts (1, Scheme 1). Initial results in this area focused on optimizing the synthesis of ruthenium boryl complexes and the exploration of the resulting reactivity of these complexes in hydroboration and C–H functionalization reactions. Replacement of the ligand-based O–H group with an O–B group (2) has led to a complex that is reactive in the hydroboration of aldehydes, ketones, and imines (published in Organometallics, 2009 and discussed in the previous progress report). Replacement of the metal-based Ru–H with a Ru–B substituent (3) results in a complex that functionalizes C-H bonds alpha- to oxygen of an ether substrate selectively over alternative terminal C-H bonds. The resulting boronate esters are prone to decomposition and could not be isolated. The novel synthesis and properties of complex 3 were recently reported (published in Organometallics, 2010).
Scheme 1. Boron-Substituted Analogues of the Shvo Hydrogenation Catalyst
Synthesis of ruthenium boryl complexes related to complex 3 (Scheme 1) was achieved by the activation of bis(catecholato)diboron in the presence and absence of a phenol. The addition of bis(catecholato)diboron to ruthenium dimer 4 provided RuBcatOBcat complex 6 in 70% NMR yield (Scheme 2). The O–B bond of complex 6 was highly labile, precluding the isolation of the pure complex. Addition of 4-methoxyphenol to complex 6 resulted in selective cleavage of the O–B bond to generate RuBcatOH complex 7. Under these conditions complex 7 was isolated in 51% yield. Alternatively, conducting the reaction between dimer 4 and bis(catecholato)diboron in the presence of 4-methoxyphenol provided complex 7 in 74% NMR yield. Isolation of complex 7 from this reaction provided a 30% yield. The isolation of 7 was hindered by the formation of a ruthenium hydride impurity. The removal of this impurity resulted in decreased yields. An X-ray crystal structure of complex 7 confirmed the anticipated structure and regiochemistry. Please see additional details in the publication (Organometallics, 2010).
Scheme 2. Synthesis of Ruthenium Boryl Complexes
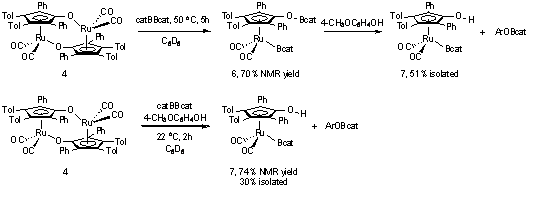
The synthesis of additional ruthenium
boryl complexes has been explored. The activation of B2cat2
shown in Scheme 2 provides low yields for the activation of B2pin2.
The application of pinacolato-substituted boronate esters to metal-ligand
bifunctional borylation reactions is desired because these complexes typically
provide products that are less prone to decomposition. Toward this end, we have
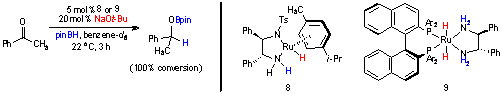
begun to explore the synthesis of boron-substituted analogues of Noyori's transfer hydrogenation (8) and hydrogenation (9)
catalysts (Scheme 3). Initially, hydroboration using these complexes was
explored to help us determine the reactivity of these complexes toward reactive
boron sources. The hydroboration of ketones using these asymmetric complexes
was also desired because the sterically congested boronate ester was expected
to increase the enantioselectivity in hydroboration reactions. To test this
hypothesis, the hydroboration of acetophenone was examined using a catalytic
amount of each Noyori catalyst. In both cases, the resulting hydroboration
product was racemic (determined by hydrolysis of O–B bond and analysis using
chiral GC).
Scheme 3. Ruthenium-Catalyzed Hydroboration of Acetophenone
Control reactions were used to
determine why the hydroboration reaction was completely unselective. The
hydroboration of acetophenone was examined in the absence of a transition metal
catalyst. The hydroboration was found to be catalyzed by NaOt-Bu rather than by Noyori's catalysts (eq 1), which accounts for the lack of
asymmetric induction. Spectroscopic studies were used to study the active
hydroboration complex. Using this information, with the observed reactivity, a
catalytic cycle was proposed (Scheme 4). These results, along with the scope of
the hydroboration reaction are the topic of a manuscript that is currently
being prepared for submission.
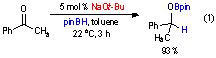
Scheme 4. Proposed Catalytic Cycle for Alkoxide-Mediated
Hydroboration of Ketones

Due to the challenges faced
with the Shvo and Noyori complexes in developing highly reactive borylation
catalysts, we shifted our attention to metal–ligand bifunctional borylation
catalysts that can easily access coordinatively unsaturated electronic states.
Rh, Ir, and Ru complexes with diamine ligands were initially chosen based on
the known role of these complexes in metal-catalyzed borylation reactions. We
have preliminary results that support this concept using iridium-catalyzed
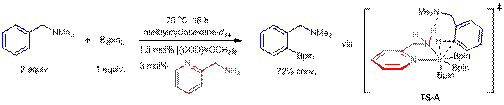
arene C–H borylation. After screening a range of diamine ligands, picolylamine was found to be a reactive ligand in the ortho
borylation of the arene C–H bond of N,
N-dimethylbenzylamine
(Scheme 5). At 70 °C, 72% conversion (with respect to B2pin2)
to the ortho borylation product was observed. Running
a similar reaction under known non-directing conditions (using 4,4'-di-tert-butylbypyridine)
led to <10% conversion, with an unselective mixture of isomers. The reaction
is believed to proceed via a hydrogen bond between the N–H bond of the ligand
and the basic amine of the substrate, leading to an optimal transition state
for ortho C–H borylation (TS-A).
Scheme 5. Ligand-Directed C–H Borylation
In summary, the synthesis of boron-substituted analogues of metal–ligand
bifunctional catalyst have provided complexes that are active as either
hydroboration catalysts or C–H borylation complexes.
