AmericanChemicalSociety.com
Reports: G1 47565-G1: Development of Carbon-Carbon Bond Activation for Organic Synthesis
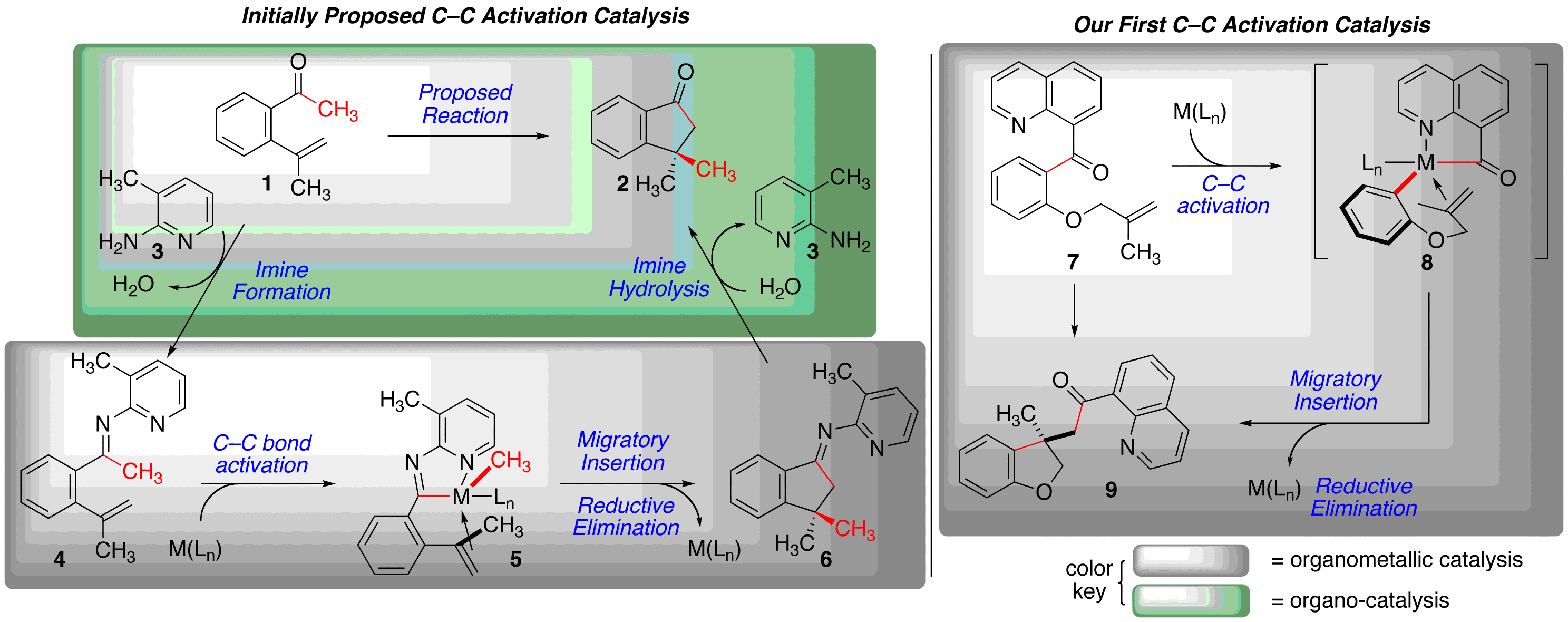
Funding from the ACS-PRF has allowed us to develop a research program in catalytic C–C s-bond activation and functionalization. This chemistry is rare, with most examples prior to our work using strained C–C s-bonds. Our initial proposal was to develop activation of C–C bonds adjacent to ketones to explore non-traditional reactivity from this standard functional group. Our initially proposed research was aimed at activating simple ketones by using the principles of organocatalysis to form the corresponding ketimine and performing organometallic catalysis to functionalized the C–C bond via chelation-assisted bond activation (Scheme 1, left). Our initial work in this area proved somewhat difficult. After identifying that the initial ketimine formation step was a problem step, we made the early tactical decision to move examine feasibility of the organometallic catalysis portion. In this mode, the activation and functionalization takes place when a chelating heteroatom is embedded in the ketone substrate (7 to 8, via 9, Scheme 1, right). We have termed this net addition of a carbon and acyl substituent across an alkene "carboacylation."
Scheme
1: Our initially proposed reaction, and
our first successful C–C activation chemistry.
We
have explored this carboacylation reaction of alkenes in inter- and
intramolecular contexts. This work
has been published and the publications added to the ACS-PRF web reporting
system. The work was summarized in
our prior progress report, and I will not discuss it further here. Just
before the last reporting period, an undergraduate student, Michael Capp,
garnered some important preliminary data on intramolecular carboacylation of
alkynes via C–C bond activation.
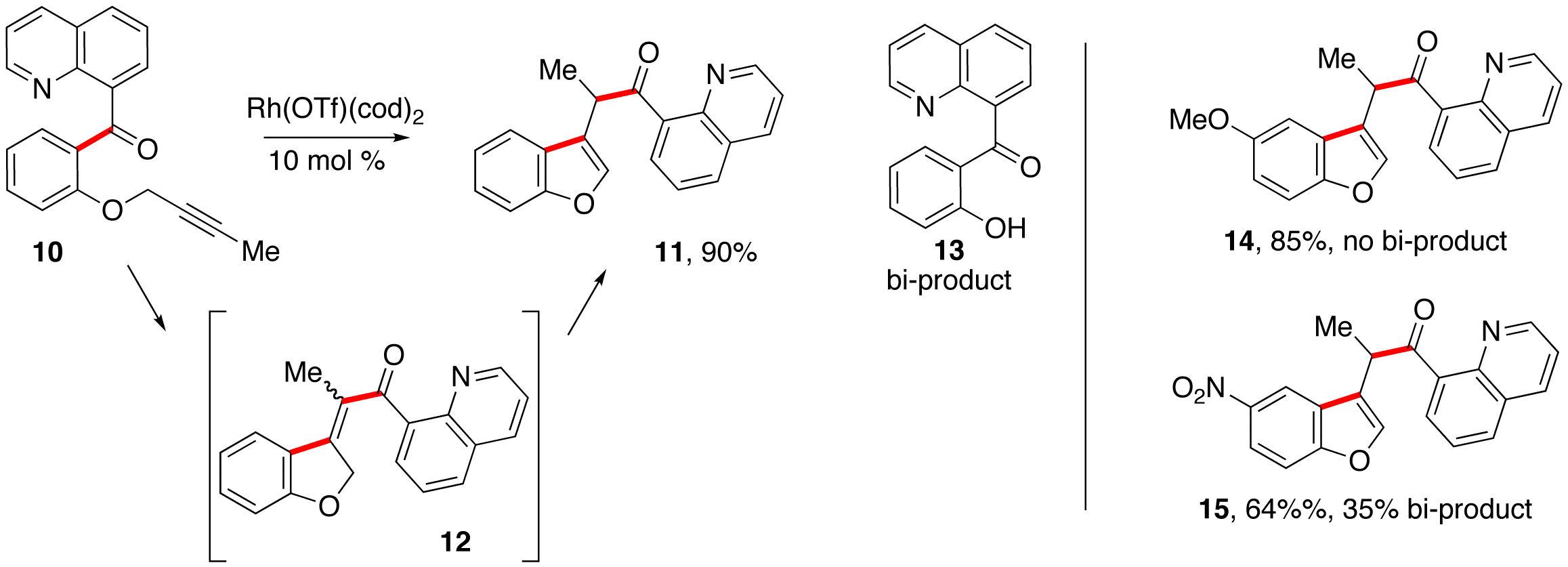
We were initially concerned that salicylaldehyde-derived 8-acyl
quinolines containing propargylic phenolic ethers would be prone more prone to
the rhodium-mediated ether cleavage (to 26,
Scheme 2) that was occasionally observed in alkene carboacylation. After examining several reaction
conditions, Michael Cap was able to identify a product resulting from alkyne
carboacylation. Although we were
initially challenged by competitive ether cleavage, Michael Wentzel and Ashley
Dreis followed up on these results, showing this bi-product could be avoided in
most cases by judicious choice of Rh catalyst and solvent. Also, they showed that a wide variety
of functional groups are tolerated in the reaction, and that the propargyl
either cleavage problem was more pronounced when the phenolic component was
substituted with electron-withdrawing groups. (Scheme 2) The substrate scope and substituent
effect study is complete, and we expect to submit a manuscript on this work in
the next few weeks.
Scheme
2: Alkyne carboacylation
Also
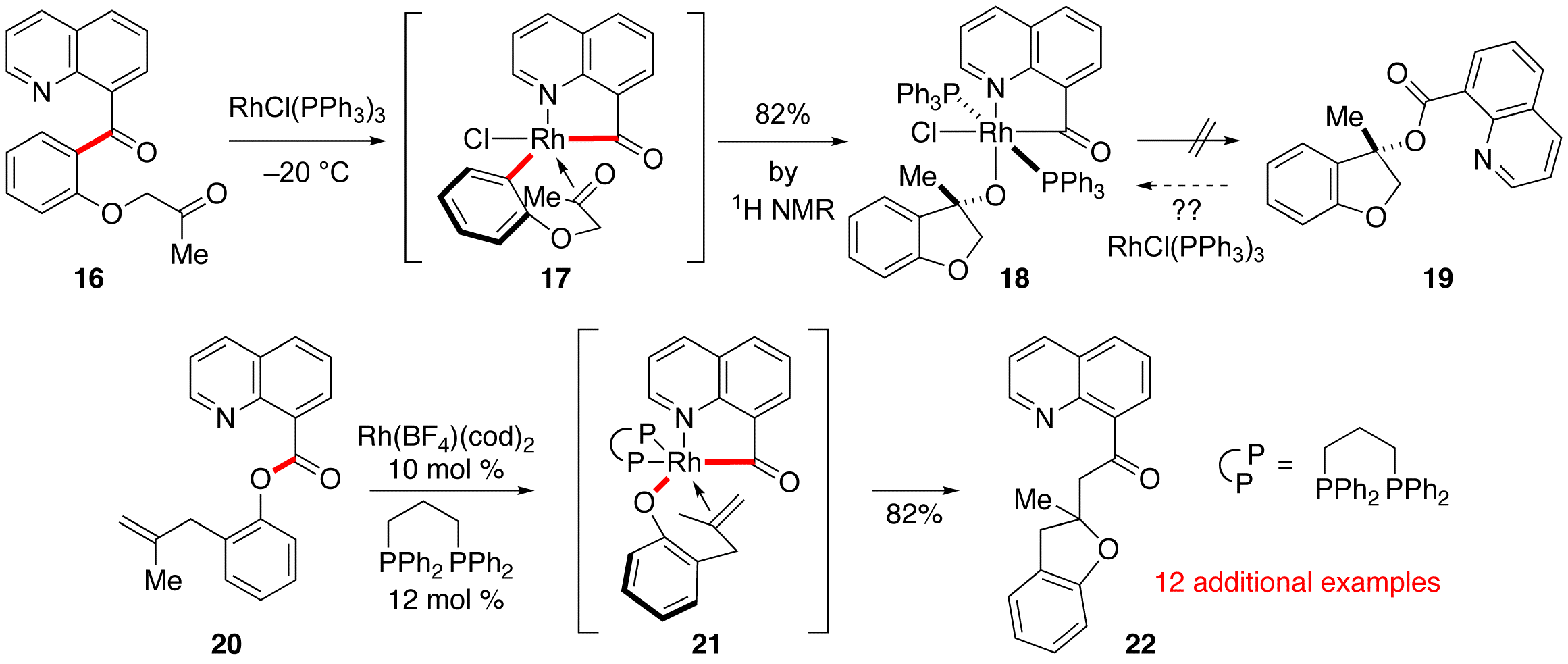
at the end of the previous reporting period, graduate student T. Giang Hoang
had initially started to probe the potential for carboacylation of ketones via
C–C activation. The outlined
experiments were along the lines alkene carboacylation, but using the directing
group to one ketone for activation and addition across the other. Giang typically saw low conversion in
this attempted experiments, so he attempted a reaction with a stoiciometric
amount of Wilkinson's catalyst.
Surprisingly, we isolated a stable rhodium-phosphine complex, and
tentatively assigned the structure as 32 based
on spectroscopy. Even more
surprisingly, the complex forms in 82% yield (by 1HNMR) at
temperatures as low as –20 °C! Giang's results lead us to question
whether the final proposed C–O reductive elimination was actually
feasible in this system. Phrased
another way, we wondered if catalytic quinoline-directed C–O activation
of esters would be a possible (such as 33 to 32), and if so,
would it allow an entry into "oxy-acylation" of alkenes. Giang undertook a study into this
reaction manifold and has recently garnered some promising catalytic results,
showing TON up to 8 for intramolecular oxyacylation. Giang has explored the substrate scope of this reaction and
his results have been submitted for publication. We will report via the ACS-PRF's web reporting system once
the work is published.
Scheme
3: Attempted Ketone Carboacylation and
C–O Activation allows "Oxy-Acylation" of Alkenes
At
the end of my previous report, I stated that we would return to the initially
proposed 2-amino pyridine system, now armed with the knowledge of what
C–C activation can accomplish with 8-acyl quinolines. A study of our initial failed attempts
at carboacylation of alkenes using these organic co-catalysts revealed that
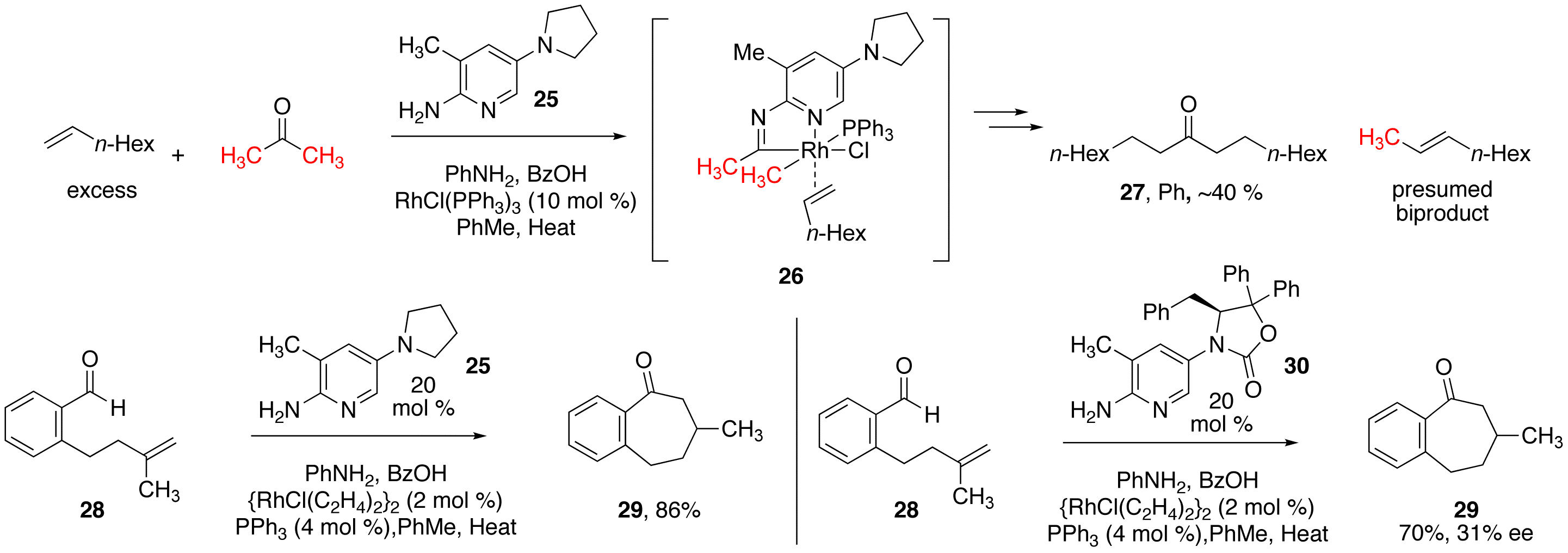
imine formation was particularly slow (as determined by in-situ NMR). Thinking to make the 2-amino-picoline a
more potent nucleophile, graduate student Evgeny (Eugene) Beletski and post-doc
Sudheer Chava examined a series of 2-aminopyridines substituted with additional
electron donor groups. Among those
examined, the structures containing a 5-amino group were most effective at
promoting imine formation.
Scheme
4: New 2-aminopyridines for C–C and
C–H Bond Activation Reactions Armed
with this knowledge, we examined the reaction of acetone (as proposed in our
initial application) with various alkenes under the action of rhodium catalysts
and our more reactive 2-aminopicoline 25
(Scheme 4). Unfortunately, only
alpha olefins have thus far been induced to react in this system. We can convert acetone into higher
ketones. This reaction only works
with our more electron rich aminopicoline additives. We are currently working to expand the system into more
complex alkenes and ketones, and will re-examine intramolecular alkene carboacylation
as we find more reactive catalyst mixtures. Concurrently,
we became interested in 2-amino-picoline/Rh catalyzed hydroacylation (Scheme 4,
bottom). Using some of the more
active picoline derivatives, we have been able to achieve the intramolecular
hydroacylation reaction to form cycloheptanones (28 29), a class
of molecules that are traditionally quite challenging to access via
hydroacylation. Taking another cue
from our original proposal, we examined chiral non-racemic 2-aminopyridine derivatives
for asymmetric induction in these hydroacylation reaction. Although not yet synthetically useful,
our preliminary results with the 2-aminopicoline derivative 30 are highly encouraging. Funding
from the ACS-PRF in the Type G award has been instrumental in garnering the
initial results that my career will no doubt be exploring for some time. Please accept my sincere thanks for
that support over the last two years. Word
Count: 973