AmericanChemicalSociety.com
Reports: B4 48565-B4: Solvent Effects in Supercritical Fluids
We are moving beyond the developmental stages of defining how supercritical fluids such as xenon, ethane, carbon dioxide, and trifluoromethane can be used as tools to study solvent effects in photophysical, photochemical, and chemical phenomena. We take advantage of the fact that operation above the critical temperature precludes the complication of phase change when the process of interest is studied as a function of pressure at constant temperature. We thereby vary the bulk-density-dependent properties such as dielectric constant, refractive index, and viscosity. A complementary view is that we also vary the local solvent number density at the solute in a predictable way. In so doing, we study solvent effects without changing the solvent, addressing both bulk effects and discrete local effects. Our method includes quantum chemical calculation and molecular dynamics simulation to define the system, and experiment to measure the behavior of the system. The developmental stage of our work has been devoted to correlation of these two computational methods with experimental observation.
In this, the second year of PRF funding, we used these funds as summer salary support for two undergraduate students, Chaim Rube and Jonathan Schwab. Our first year of funding and university matching funds allowed us to purchase and house a combined steady-state and time-resolved spectrofluorometer, which was placed into operation towards the end of our first year of PRF funding. The time-resolved component of this instrumentation allows us to measure excited state lifetimes on the picosecond time scale. The light sources and the emission under supercritical conditions are coupled to a four-windowed, high-pressure, stainless-steel vessel via fiber optics and liquid light guides. We have devoted much of our experimental effort in this second year to overcoming the non-trivial obstacles that accompany sensitive measurements in unusual media under unusual conditions. Jonathan Scwab has made substantial contributions to our concomitant steady-state absorbance work.
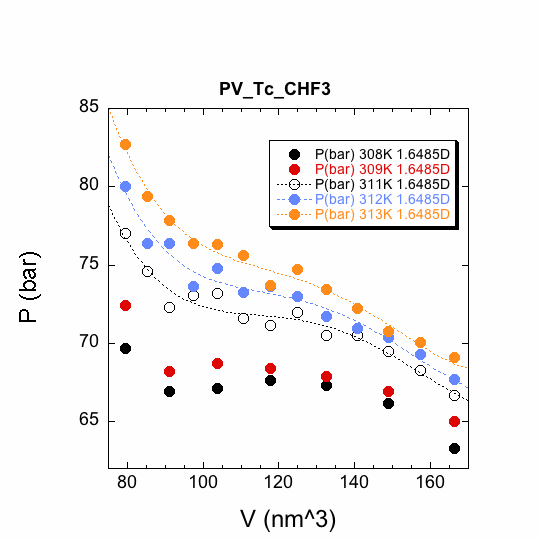
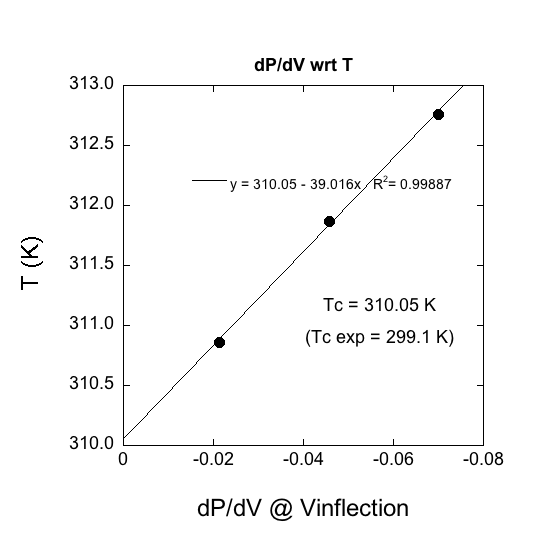
Our most tangible progress has been the development of an all-atom trifluoromethane model for classical molecular dynamics simulations. We believe that this is the best all-atom model to date; it reproduces the 1.65 D dipole moment and incorporates only bond distance contraints. Bond angles are defined by harmonic potentials. A molecular dynamics annealing experiment with one pyrazine and one trifluoromethane gives a structure that is remarkably close in energy and configuration to a quantum chemically geometry optimized structure at the MP2/6-311++G(d,p) level. The student Chaim Rube ran simulations to define isotherms close to the critical temperature for a system of 500 trifluoromethanes and one pyrazine according to a standard Lennard-Jones potential. The three isotherms (311, 312, and 313 K) above the critical temperature were fitted with fourth-order polynomials (Figure 1). Solutions to the first derivatives at the inflection points were plotted against temperature; the T-intercept gives the critical temperature for these simulation conditions (Figure 2).
Figure 1. Isotherms for
our Lennard-Jones simulation of all-atom trifluoromethane used to determine the
critical temperature, 310.5 K, which is within about 5 percent of the
experimental value, 305.3 K.
Figure 2. Slopes of the
inflection points versus temperature for isotherms close to and above the
simulation critical temperature for our Lennard-Jones simulation of trifluoromethane.
Assumption of linearity very
close to the critical temerature gives the critical temperature 310.05 K. We
are grateful for support from the Petroleum Research Foundation, and look
forward to acknowledging this support in forthcoming publications.