AmericanChemicalSociety.com
Reports: AC3 48546-AC3: Probing the Articulation Between Electron Transfer and Bond Cleavage Using Early Metal Complexes of Redox-Active Phenoxides
The chemical heart of a fuel cell is a catalyst that can mediate between the worlds of single-electron outer-sphere transfer relevant to passing current, and that of multi-electron inner-sphere oxidations and reductions involved in making and breaking the chemical bonds of fuel and oxidant. This research explores a new way of mediating between these two worlds, using a metal complex where the organic ligand undergoes one-electron redox chemistry, and the metal atom holds the substrate in physical and electronic contact with the ligand and facilitates its changes in bonding.
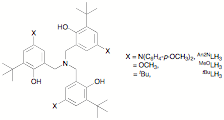
New diarylaminoaryloxide ligands and their complexes
We recently published a description of the preparation of new tripodal aminetriphenol ligands LH3 containing p-diarylaminophenoxide groups. These ligands have the added advantages of conferring excellent water-stability on their early metal complexes, and of allowing an open coordination site in complexes LTiX. Both these features are likely to be important in allowing catalytic applications, particularly under ambient (i.e., wet) conditions. We have made a di-(4-methoxyphenyl)amino-substituted tripodal ligand An2NLH3, and have compared its properties with the methoxy-substituted MeOLH3 that we recently prepared and the well-known tert-butyl substituted tBuLH3.
Cyclic
voltammetry studies of the six-coordinate LTi(acac) complexes illuminate the
effect of the diarylamino substituent on the redox properties of the metal
complexes, with An2NLTi(acac) showing reversible oxidations for each
of the three arms, separated by about 150 mV from each other. (In fact, since each of the arms also
shows a second one-electron oxidation at about 700 mV higher potential than the
first, An2NLTi(acac) can effectively store up to 6 oxidizing equivalents.) Thus, by moving the locus of oxidation
farther from the metal, An2NLTi(acac) minimizes the difference in
potential between successive oxidations.
At the same time, the retention of a modest electronic coupling between
the aryloxide groups suggests that there is enough electronic communication
between the radical centers and the titanium to allow ligand oxidation to
affect the chemistry of other groups coordinated to the metal center. We
are also exploring the use of these tripodal ligands to support oxometal
fragments where the oxo group can bind a proton with concomitant one- or
two-electron redox changes at the ligand to allow proton-coupled electron
transfer reactions. Such reactions
typically involve redox changes at metal centers coupled with proton transfer
to ligands; our use of ligands to carry out both changes is unusual (though
precedented, for example, by oxometal complexes of porphyrin radical cations
that likely carry out oxidations in cytochromes P450). So far, we have found that LMo(O)2–
and LW(O)2– salts can be prepared directly from
molybdate and tungstate under mild conditions, which emphasizes their
hydrolytic stability. Compounds
with the tert-butyl and
methoxy-substituted ligands may be reversibly oxidized by one (but only one)
electron, while the
dianisylamino-substituted ligands can be oxidized reversibly by up to
three electrons (though the first oxidation is much more facile than the
subsequent ones, in contrast to the behavior of the titanium compounds). All the anions can be protonated
reversibly by mild acids (e.g., saccharin). Combining the pKa and E¼ data indicates that the
neutral species LMo(O)(OH) and LW(O)(OH) have O–H bond dissociation free
energies ranging from 81 kcal/mol to 64 kcal/mol. This is an enormous range for a series of essentially
isosteric and isoelectronic compounds, and we are currently exploring their
PCET reactions to see how well the ligand-based redox changes couple to
bond-breaking or ‑forming reactions at the metal oxo group.
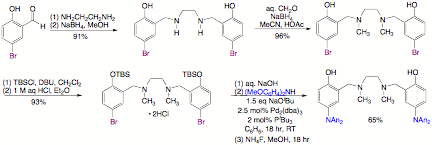
We
have also explored tetradentate, dianionic N2O2 ligands
of the salan type, which are prepared efficiently on a large scale. N,N'-bis(5-bromo-2-hydroxybenzyl)-N,N'-dimethylethylenediamine
is prepared through two successive reductive aminations from commercially available
5-bromosalicylaldehyde. Protection
of the phenols as tert-butyldimethylsilyl ethers, followed by Hartwig-Buchwald
amination using bis(4-methoxyphenyl)amine and desilylation, affords the
corresponding salan ligand with two redox-active di(4-methoxyphenyl)aminoaryloxides.
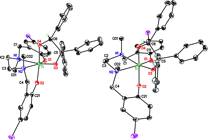
We
have prepared and characterized a wide variety of complexes of the redox-active
An2Nsalan (and the redox-resistant Brsalan, as controls) with
ancillary ligands such as isopropoxide, chloride, and a- and b-hydroxyacids. The ligand appears to be rather
flexible, adopting symmetrical cis-a or unsymmetrical cis-b
arrangements depending on the nature of the ancillary ligands in a way that can
be explained on the basis of those ligands' s-
or ¹-donating abilities. Most
remarkable are complexes of 1,1,2-triphenylethanediolate, which we have
characterized crystallographically in both cis-b, trans N-methyl and a very unusual cis-b, cis
N-methyl geometry (both are observed in
solution as well, with the former being the major isomer).
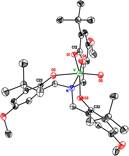
We
have also explored complexes of a novel
2,2'-biphenol-3,3'-di-(2-hydroxyphenylimine) ligand as a potentially
multinucleating redox-active phenolate.
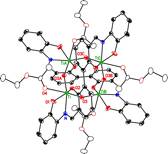
While the ligand forms stable complexes of interesting architecture (e.g.,
the tetratitanium complex below), its redox properties are not promising.
Linking Ligand Oxidation and Substrate Bond Cleavage The 1,1,2-triphenylethanediolate complexes of
(salan)Ti(IV) have proven to be outstanding platforms for assessing the ability
of ligand oxidation to induce bond breaking in a bound substrate molecule. Remarkably, while the salan complexes
are chemically stable for prolonged periods even at elevated temperatures, they
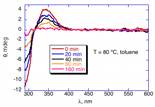
are not optically stable:
racemization takes place at appreciable rates at 80 ¼C and can be
measured quantitatively by circular dichroism spectroscopy. Mechanistic observations (low solvent
effect, low sensitivity to salan donor ability, positive entropy of activation)
are all consistent with this reaction taking place by simple C–C bond
homolysis.
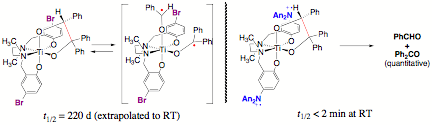
In
contrast to the very slow, reversible C–C bond cleavage that is accessed
thermally for the neutral compounds, outer-sphere oxidation of (An2Nsalan)Ti(OCHPhCPh2O),
for example by ferrocenium or silver(I), results in rapid quantitative loss of
benzophenone and benzaldehyde.
Detailed quantitative kinetic measurements are in progress, but it is
already clear that the half-life for bond cleavage in the oxidized compound is
< 2 min. Thus, aryloxide
oxidation results in a > 105 rate acceleration for bond cleavage,
indicating that the strategy of coupling ligand oxidation with inner-sphere
reactions of substrates is a potentially viable approach to mediating redox
processes of relevance to fuel cells.